Received: February 2018
DOI 10.17677/fn20714807.2018.01.05
Fluorine Notes, 2018, 116, 9-10
Synthesis of 5-nitro-pyrazole triflones via [3+2] cycloaddition reaction and its application for potential insecticide
Pulakesh Das,1 Hiroto Uno, 1 Etsuko Tokunaga, 1 Yuji Sumii 1 and Norio Shibata1,2*
1Department of Nanopharmaceutical Sciences, and Department of Life Science and Applied Chemistry,
Nagoya Institute of Technology, Gokiso, Showa-ku, Nagoya 466-8555, Japan, E-mail: nozshiba@nitech.ac.jp; TEL; +81-52-735-7543
2Institute of Advanced Fluorine-Containing Materials, Zhejiang Normal University, 688 Yingbin Avenue, 321004 Jinhua, China
Abstract: Synthesis of 5-nitro-pyrazole triflones 6 was achieved via [3+2] cycloaddition reaction of 2-diazo-1-phenyl-2-((trifluoromethyl)sulfonyl)ethan-1-one (3) and α-bromonitrostyrene derivatives 7 under basic conditions in moderate to good yields. An agrochemically attractive 5-amino-N-pyrimidinyl-pyrazole triflone 2a was successfully prepared from 5-nitro-pyrazole triflone 6c in two steps.
Key words: 5-nitro-pyrazole triflones, α-bromonitrostyrene, [3+2] cycloaddition
Introduction: Pyrazoles are often found as an integral part of biologically active molecules [1]. In particular, pyrazoles with aryl or heteroaryl substituents at N-1 position and a free NH2 group at C-5 position have gathered much attention because of their promising pharmaceutical [2] and agrochemical [3] properties. More precisely, 1-arylpyrazoles having substituents like alkyl, thioalkyl, acyl or cyano- group at the C-3 and C-4 positions exhibit potent insecticidal activities [4]. Amongst the pyrazole-type pesticides, Fipronil® (5-amino-1-(2,6-dichloro-4-trifluoromethylphenyl)-4-trifluoromethanesulfinyl-1H-pyrazole-3-carbonitrile) is a highly effective broad-spectrum insecticide and veterinary medicine to get rid of fleas and ticks [5] (Figure 1). However, the use of Fipronil® is the matter of discussion due to the recent food safety incident [6]. Thus, the development of safer derivatives of Fipronil® is of great importance. Bayer CropScience AG has filed patents on the sulfur-derivatized pyrimidinyl-pyrazol-5-amines 1 which showed potent activities as insecticides and/or parasiticides [3a,7]. Inspired by their patents, we are interested in triflyl (trifluoromethanesulfonyl, SO2CF3) variants (triflones) 2 as potential pesticides.
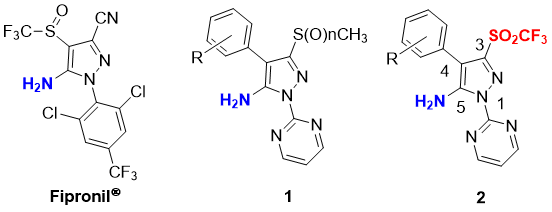
Figure 1. Fipronil® and its biologically attractive derivatives containing aryl or heteroaryl substituent at N-1 position and a free NH2 group at C-5 position.
The introduction of fluorine(s) and fluorinated functional groups into organic compounds is one of the effective strategies to improve/alter biological activity and metabolic stability of original molecules [8]. In the case of our target 5-amino-pyrazole triflones 2, the incorporation of a lipophilic triflyl group (π = 0.55) to C-3 position in 2 could potentially perturb the chemical, physical and biological properties of the parent heterocyclic 5-amino-pyrazoles. Besides, the triflone variants 2 are also expected to have a reduced basicity of the amine moiety in 2 due to the strong electron withdrawing effect by the triflyl group (σm = 0.79, σp = 0.93) [9]. For decades, our group has been focusing on the development of novel shelf-stable reagents for fluoro-functionalization reactions [10,11]. One such kind of unique compounds is 2-diazo-1-phenyl-2-((trifluoromethyl)-sulfonyl)ethan-1-one (3) [12]. The compound 3 was originally developed by us as a regent for electrophilic trifluoromethylthiolation reactions. A wide variety of nucleophiles such as enamines, indoles, β-ketoesters, and pyrroles are nicely trifluoromethylthiolated by 3 in the presence of a copper catalyst under mild conditions [12a]. Aromatic coupling reaction is also available by using 3. On the other hand, 3 also acts as a powerful building block for the preparation of variety of triflones such as β-lactam triflones [12c]. Recently, we have reported the synthesis of pyrazole triflones 4 via [3+2] cycloaddition of 3 with nitrostyrenes 5 (Scheme 1a) [12d]. In the presence of NaOMe, reagent 3 generates a reactive anionic triflyl-diazomethane species A in situ to react with nitrostyrenes 5. As an extension of our research work on heterocyclic triflones [12], we herein report the novel strategy for the preparation of 5-amino-N-pyrimidinyl-pyrazole triflones 2. The important precursors to access 2 are the 5-nitro-pyrazole triflones 6, which can be synthesized by the reaction of 3 with α-bromonitrostyrenes 7 under basic conditions (Scheme 1b). The selective elimination of the bromo group rather than nitro group in 7 during the cyclization step to suppress the formation of 5-bromo-pyrazole triflones 8 is the key for the success [13]. Substrate scope of 7 for the preparation of 6 was examined. Subsequently, the 5-nitro-pyrazole triflone 6c was successfully transformed to the targeted 5-amino-N-pyrimidinyl-pyrazole triflone 2a via two steps in moderated yield (Scheme 1b).
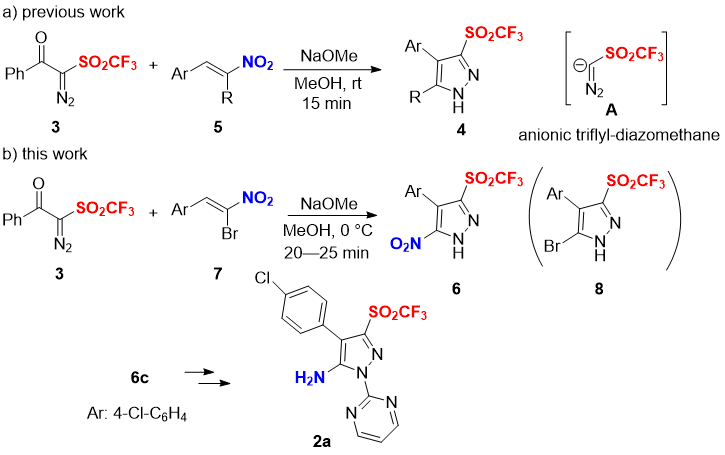
Scheme 1. a) Synthesis of pyrazole triflons 4 via [3+2] cycloaddition of 3 with nitrostyrenes 5 (previous work); b) Selective synthesis of 5-nitro-pyrazole triflons 6 and transformation of 6c to 5-amino-N-pyrimidinyl-pyrazole triflone 2a (this work).
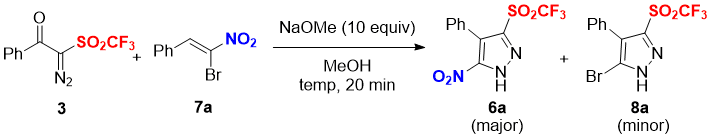
Based on our previous work [12d], we initiated to optimize the reaction temperatures for the reactions between 3 and α-bromonitrostyrene 7a in the presence of 10 equiv of NaOMe in MeOH (Table 1). Desired 5-nitro-pyrazole triflone 6a was obtained in 62% yield along with 5-bromo-pyrazole triflone 8a in 17% yield at 0 °C for 20 min (entry 1). While, decreasing the reaction temperature to –10 °C, did not affect the yield and products distribution (entry 2). Increasing the amount of 3 from 1.2 to 1.5 equiv, slightly increased the yields of 6a to 64% and 8a to 19% (entry 3) were detected, whereas lowering the temperature with 1.5 equiv of 3 did not affect the formation of nitro product 6a with slightly increased the yield of bromo 8a to 21% (entry 4). Thus, the combination of 3 (1.5 equiv) and α-bromonitrostyrenes 7 (1.0 equiv) in the presence of NaOMe (10.0 equiv) in MeOH at 0 °C for 20 min, was selected as the optimized reaction condition (entry 3).
Table 1. Optimization of [3+2] cycloaddition reactiona
|
entry |
3 (equiv) |
7a (equiv) |
temp. (°C) |
yield (%) |
|
|
6a |
8a |
||||
|
1 |
1.2 |
1.0 |
0 |
62 |
17 |
|
2 |
1.2 |
1.0 |
–10 |
63 |
20 |
|
3 |
1.5 |
1.0 |
0 |
64 |
19 |
|
4 |
1.5 |
1.0 |
–10 |
64 |
21 |
aReaction conditions: Experiments were performed with 3 (1.2 or 1.5 equiv), 7a (1.0 equiv), NaOMe (10 equiv), MeOH (1.0 mL) at given temperature for 20 min.
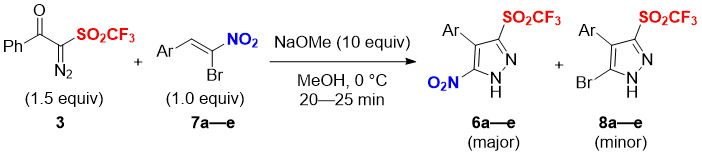
With the optimized reaction conditions in hand, the substrate scope of the [3+2] cyclization reaction was explored as shown in Table 2. All the reactions proceeded smoothly to furnish the desired 5-nitro-pyrazole triflones 6b—e as major products in good yields with similar selectivity as 7a. Electron donating 4-Me-C6H4 substituted substrate 7b provided the product in similar yield and selectivity (6b: 67%, 8b: 23%). The halogen substituted α-bromonitrostyrenes (7c: 4-Cl-C6H4; 7d: 3-Br-C6H4; 7e: 4-F-C6H4) provided the corresponding 5-nitro-pyrazole triflones 6 with higher yields (6c: 77%; 6d: 76%; 6e: 69%) along with corresponding bromo by-products 8 (8c: 15%; 8d: 18%; 8e: 17%) (Table 2).
Next, we examined the synthesis of 5-amino-N-pyrimidinyl-pyrazole triflone 2a. The reduction of nitro group in 6c by hydrogenation in the presence of Pd-C in MeOH gave the 5-amino-pyrazole triflone 9, followed by the nucleophilic addition to 2-chloropyrimidine in the presence of NaH under heating condition to afford the potential biological active 5-amino-N-pyrimidinyl-pyrazole triflone 2a in 35% yield for two steps (Scheme 2).
Table 2. Scope of the reaction of diazotriflone 3 with α-bromonitroalkenes 7a-ea
|
entry |
7 |
Ar |
Yield (%) |
|
|
6 |
8 |
|||
|
1 |
7a |
Ph |
6a: 64 |
8a: 19 |
|
2 |
7b |
4-Me-C6H4 |
6b: 67 |
8b: 23 |
|
3 |
7c |
4-Cl-C6H4 |
6c: 77 |
8c: 15 |
|
4 |
7d |
3-Br-C6H4 |
6d: 76 |
8d: 18 |
|
5 |
7e |
4-F-C6H4 |
6e: 69 |
8e: 17 |
aExperiments were performed with 3 (1.5 equiv), 7a-e (1.0 equiv) and NaOMe (10 equiv) in dry MeOH at 0 °C for 20–25 min.
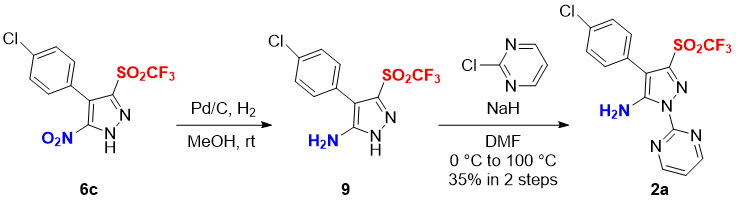
Scheme 2. Synthesis of 5-amino-N-pyrimidinyl-pyrazole triflone 2a.
In conclusion, we have disclosed the synthesis of 5-nitro-pyrazole triflones 6 via [3+2] cycloaddition reaction of 2-diazo-1-phenyl-2-((trifluoromethyl)sulfonyl)ethan-1-one 3 and α-bromonitrostyrene derivatives 7 under basic conditions in moderate to good yields. The transformation of 5-nitro-pyrazole triflone 6 to an agrochemically attractive 5-amino-N-pyrimidinyl-pyrazole triflone 2a was successfully demonstrated in two steps. The synthesis of a series of 5-amino-N-pyrimidinyl-pyrazole triflones and their evaluation of the insecticide property are underway.
Experimental
All reactions were performed in oven-dried glassware under positive pressure of nitrogen or argon unless mentioned otherwise. Solvents were transferred via syringe and were introduced into reaction vessels though a rubber septum. All reactions were monitored by thin-layer chromatography (TLC) carried out on 0.25 mm Merck silica gel (60-F254). The TLC plates were visualized with UV light and KMnO4 in water/heat. Column chromatography was carried out on columns packed with silica gel (60N spherical neutral size 40-50 µm) for flash column chromatography. The 1H NMR (300 MHz), 19F NMR (282 MHz), and 13C NMR (126 MHz) spectra for solution in CDCl3, CD3OD were recorded on Varian Mercury 300, and Bruker Avance 500 NMR spectrometers. Chemical shifts (δ) are expressed in ppm downfield from TMS (δ = 0.00) or C6F6 [δ = −162.2 (CDCl3)] as an internal standard. Mass spectra were recorded on a SHIMAZU LCMS-2020 (ESI-MS). High resolution mass spectrometric measurements were recorded on a Waters Synapt G2 HDMS (ESI-MS), a Waters GCT premier (EI-MS). Solvents CH3CN, CH2Cl2, DMF were dried and distilled before use. 2-Diazo-1-phenyl-2-((trifluoromethyl)-sulfonyl)ethan-1-one (3) [12c] and (Z)- α-bromonitrostyrenes 7a—e [14] were prepared according to the known procedures.
Typical procedure for the synthesis of 5-nitro-4-aryl-pyrazole triflon (6) and 5-bromo-4-aryl-pyrazole triflon (8) from 3 and (Z)-1-(2-bromo-2-nitrovinyl) benzene (7) (Procedure A)
To a stirred solution of (Z)-α-bromonitrostyrenes 7 (0.1 mmol, 1.0 equiv) and 2-diazo-1-phenyl-2-((trifluoromethyl)-sulfonyl)ethan-1-one (3) (0.15 mmol, 1.5 equiv) in dry MeOH (1.0 mL), NaOMe (10.0 equiv) was added at 0 °C and the resulting mixture was stirred for 20-25 minutes under Ar atmosphere (progress of the reaction was monitored by TLC). After completion, the reaction mixture was concentrated under reduced pressure. Water was added and neutralized the solution with 5% HCl and extracted with ethyl acetate. The combined organic phase was washed with brine and dried over Na2SO4, and the mixture was evaporated in vacuo. The crude product was purified by flash column chromatography (DCM/MeOH = 10:1) to obtain the desired substituted 5-nitro-4-phenyl-3-((trifluoromethyl)sulfonyl)-1H-pyrazole 6 and 5-bromo-4-phenyl-3-((trifluoromethyl)sulfonyl)-1H-pyrazole 8.
5-Nitro-4-phenyl-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (6a) and 5-Bromo-4-phenyl-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (8a):
(Z)-(2-Bromo-2-nitrovinyl) benzene 7a (0.1 mmol, 0.023 g, 1.0 equiv), 3 (0.15 mmol, 0.042 g, 1.5 equiv) and NaOMe (1.0 mmol, 0.054 g, 10.0 equiv) in dry MeOH (1.0 mL) at 0 °C for 20 min provided the pure product 6a (0.0205 g, 64 %) as a pale yellow solid and 8a (0.0068 g, 19 %) as a brown solid.
6a: Mp: 313–315 °C (methanol); 1H NMR (CDCl3, 300 MHz) δ: 7.56–7.44 (m, 3H), 7.40–7.31 (m, 2H); 19F NMR (CDCl3, 282 MHz) δ: –77.96 (s, 3F); 13C NMR (CD3OD, 126 MHz) δ: 151.51, 138.17, 131.37, 130.22, 128.86, 127.58, 125.48, 120.81 (q, J = 324.8 Hz); IR (KBr): 3581, 3334, 3066, 1963, 1673, 1560, 1506, 1378, 1232, 1110, 921 cm-1; HRMS (ESI–TOF): calcd for C10H5F3N3O4S [M–H]– 319.9953; found 319.9981.
8a: Mp: 152–155 °C (DCM); 1H NMR (CDCl3, 300 MHz) δ: 7.49–7.43 (m, 3H), 7.41–7.36 (m, 2H); 19F NMR (CDCl3, 282 MHz) δ: –78.30 (s, 3F); 13C NMR (CDCl3, 126 MHz) δ: 130.49, 129.92, 129.55, 129.28, 128.49, 127.82, 126.77, 119.51 (q, J = 326.0 Hz); IR (KBr): 3642, 3210, 3116, 2672, 1959, 1654, 1533, 1386, 1114, 987, 717 cm-1; HRMS (ESI–TOF): calcd for C10H6F3N2O2SBrNa [M+Na]+ 376.9183; found 376.9177.
5-Nitro-4-(p-tolyl)-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (6b) and 5-Bromo-4-(p-tolyl)-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (8b):
(Z)-1-(2-Bromo-2-nitrovinyl)-4-methylbenzene 7b (0.3 mmol, 0.0726 g, 1.0 equiv), 3 (0.45 mmol, 0.125 g, 1.5 equiv) and NaOMe (3.0 mmol, 0.162 g, 10.0 equiv) in dry MeOH (3.0 mL) at 0 °C for 25 min provided the pure product 6b (0.0677 g, 67%) as a pale yellow solid and 8b (0.0251 g, 23%) as a brown solid.
6b: Mp: 305–308 °C (methanol); 1H NMR (CDCl3, 300 MHz) δ: 7.34–7.15 (m, 4H), 2.43 (s, 3H); 19F NMR (CDCl3, 282 MHz) δ: –78.12 (s, 3F); 13C NMR (CD3OD, 126 MHz) δ: 155.17, 139.39, 139.21, 131.23, 129.18, 126.95, 125.27, 121.20 (q, J = 325.0 Hz), 21.37; IR (KBr): 3596, 3210, 3035, 1720, 1670, 1529, 1371, 1224, 1110, 995 cm-1; HRMS (ESI–TOF): calcd for C11H7F3N3O4S [M–H]– 334.0109, found 334.0108.
8b: Mp: 133–134 °C (DCM); 1H NMR (CDCl3, 300 MHz) δ: 7.29–7.25 (m, 4H), 2.41 (s, 3H); 19F NMR (CDCl3, 282 MHz) δ: –78.33 (s, 3F); 13C NMR (CDCl3, 126 MHz) δ: 139.55, 130.33, 129.72, 129.23, 127.77, 125.33, 123.79, 119.53 (q, J = 325.7 Hz), 21.56; IR (KBr): 3612, 3237, 3097, 2350, 1901, 1662, 1498, 1382, 1106, 975, 717 cm-1; HRMS (ESI–TOF): calcd for C11H8F3N2O2SBrNa [M+Na]+ 390.9340, found 390.9333.
4-(4-Chlorophenyl)-5-nitro-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (6c) and 5-Bromo-4-(4-chlorophenyl)-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (8c):
(Z)-1-(2-Bromo-2-nitrovinyl)-4-chlorobenzene 7c (0.3 mmol, 0.0787 g, 1.0 equiv), 3 (0.45 mmol, 0.125 g, 1.5 equiv) and NaOMe (3.0 mmol, 0.162 g, 10.0 equiv) in dry MeOH (3.0 mL) at 0 °C for 20 min provided the pure product 6c (0.0825 g, 77 %) as a brownish yellow solid and 8c (0.0177 g, 15 %) as a brown solid.
6c: Mp: 271–273 °C (methanol); 1H NMR (CDCl3, 300 MHz) δ: 7.38 (d, J = 8.1 Hz, 2H), 7.16 (d, J = 8.2 Hz, 2H), 3.04 (br s, 1H); 19F NMR (CDCl3, 282 MHz) δ: –78.47 (s, 3F); 13C NMR (CD3OD, 126 MHz) δ: 156.09, 139.82, 135.04, 132.99, 129.66, 128.60, 123.65, 121.26 (q, J = 325.2 Hz); IR (KBr): 3507, 3241, 3073, 2609, 1897, 1635, 1529, 1367, 1220, 1106, 848 cm-1; HRMS (ESI–TOF): calcd for C10H4F3N3O4SCl [M–H]– 353.9563; found 353.9581.
8c: Mp: 136–138 °C (DCM); 1H NMR (CDCl3, 300 MHz) δ: 7.45 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H); 19F NMR (CDCl3, 282 MHz) δ: –78.32 (s, 3F); 13C NMR (CDCl3, 126 MHz) δ: 135.73, 133.76, 131.71, 130.22, 128.75, 126.43, 125.12, 119.34 (q, J = 325.9 Hz); IR (KBr): 3619, 3131, 2923, 2327, 1540, 1459, 1390, 1110, 987, 829, 717 cm-1; HRMS (ESI–TOF): calcd for C10H4F3N2O2SClBr [M–H]– 386.8817; found 386.8824.
4-(3-Bromophenyl)-5-nitro-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (6d) and 5-Bromo-4-(3-bromophenyl)-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (8d):
(Z)-1-Bromo-3-(2-bromo-2-nitrovinyl)benzene 7d (0.3 mmol, 0.092 g, 1.0 equiv), 3 (0.45 mmol, 0.125 g, 1.5 equiv) and NaOMe (3.0 mmol, 0.162 g, 10.0 equiv) in dry MeOH (3.0 mL) at 0 °C for 20 min provided the pure product 6d (0.0917 g, 76 %) as a brownish yellow solid and 8d (0.024 g, 18%) as a brown solid.
6d: Mp: 266–268 °C (methanol); 1H NMR (CDCl3, 300 MHz) δ: 7.59 (d, J = 7.5 Hz, 1H), 7.43 (s, 1H), 7.36–7.26 (m, 1H), 7.20 (d, J = 7.4 Hz, 1H); 19F NMR (CDCl3, 282 MHz) δ: –78.54 (s, 3F); 13C NMR (CD3OD, 126 MHz) δ: 156.12, 139.87, 134.13, 133.27, 131.95, 130.31, 130.17, 123.17, 122.15, 121.26 (q, J = 325.3 Hz); IR (KBr): 3569, 3226, 3085, 2613, 1743, 1552, 1521, 1378, 1205, 1106, 840, 624 cm-1. HRMS (ESI–TOF): calcd for C10H4F3N3O4SBr [M–H]– 397.9058; found 397.9055.
8d: Mp: 115–116 °C (DCM); 1H NMR (CDCl3, 300 MHz) δ: 7.61 (d, J = 5.9 Hz, 1H), 7.54 (s, 1H), 7.40–7.31 (m, 2H); 19F NMR (CDCl3, 282 MHz) δ: –78.27 (s, 3F); 13C NMR (CD3OD, 126 MHz) δ: 134.51, 134.04, 133.04, 131.41, 130.96, 130.67, 129.47, 126.43, 122.86, 120.90 (q, J = 325.3 Hz); IR (KBr): 3667, 3139, 2938, 2373, 1556, 1459, 1375, 1106, 987, 871, 790, 655 cm-1; HRMS (ESI–TOF): calcd for C10H4F3N2O2SBr2 [M–H]– 430.8312; found 430.8305.
4-(4-Fluorophenyl)-5-nitro-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (6e) and 5-Bromo-4-(4-fluorophenyl)-3-((trifluoromethyl)sulfonyl)-1H-pyrazole (8e):
(Z)-1-(2-Bromo-2-nitrovinyl)-4-fluorobenzene 7e (0.3 mmol, 0.0738 g, 1.0 equiv), 3 (0.45 mmol, 0.125 g, 1.5 equiv) and NaOMe (3.0 mmol, 0.162 g, 10.0 equiv) in dry MeOH (3.0 mL) at 0 °C for 20 min provided the pure product 6e (0.0706 g, 69 %) as a brownish yellow solid and 8e (0.0188 g, 17 %) as a brown solid.
6e: Mp: 352–354 °C (methanol); 1H NMR (CDCl3, 300 MHz) δ: 7.26–7.23 (m, 2H), 7.18–7.07 (m, 2H), 2.20 (br s, 1H); 19F NMR (CDCl3, 282 MHz) δ: –78.65 (s, 3F), –111.50 (s, 1F); 13C NMR (CD3OD, 126 MHz) δ: 162.65 (d, J = 245.3 Hz), 155.03, 138.54, 131.98 (d, J = 8.6 Hz), 125.71 (d, J = 3.8 Hz), 122.47, 119.91 (q, J = 325.2 Hz), 113.82 (d, J = 22.0 Hz); IR (KBr): 3519, 3318, 3097, 2562, 1654, 1521, 1375, 1224, 1106, 991 cm-1; HRMS (ESI–TOF): calcd for C10H4F4N3O4S [M–H]– 337.9859; found 337.9861.
8e: Mp: 116–118 °C (DCM); 1H NMR (CDCl3, 300 MHz) δ: 7.40–7.33 (m, 2H), 7.19–7.12 (m, 2H); 19F NMR (CDCl3, 282 MHz) δ: –78.35 (s, 3F), –111.67– –111.86 (m, 1F); 13C NMR (CD3OD, 126 MHz) δ: 164.57 (d, J = 247.0 Hz), 133.89 (d, J = 8.7 Hz), 130.72, 129.47, 127.02, 125.31 (d, J = 3.6 Hz), 120.92 (q, J = 325.1 Hz), 116.10 (d, J = 22.1 Hz); IR (KBr): 3679, 3127, 2931, 2377, 1548, 1475, 1382, 1228, 1114, 983, 755, 640 cm-1; HRMS (ESI–TOF): calcd for C10H4F4N2O2SBr [M–H]– 370.9113; found 370.9113.
Synthesis of 4-(4-chlorophenyl)-1-(pyrimidin-2-yl)-3-((trifluoromethyl)sulfonyl)-1H-pyrazol-5-amine (2a)
To a solution of 4-(4-chlorophenyl)-5-nitro-3-((trifluoromethyl)sulfonyl)-1H-pyrazole 6c (0.0355 g, 0.1 mmol) in MeOH was added Pd/C (0.006 g), and the mixture was stirred at rt under H2 atmosphere (balloon) for 2.5 h. The mixture was filtered through a pad of Celite® to give the amine 9. The crude product was used for the next reaction without further purification.
The crude amine 9 was dissolved in DMF (1.0 mL). NaH (60% w/w in mineral oil, 0.0024 g, 0.1 mmol) was added to the mixture at 0 °C, and the mixture was stirred at rt for 30 min. 2-Chloropyrimidine (0.0115 g, 0.1 mmol) was added to the mixture and the mixture was stirred at 100 °C for 12 h. H2O was added to the mixture and the mixture was extracted with EtOAc. The combined organic phase was washed with brine and dried with Na2SO4, and the mixture was evaporated in vacuo. The crude product was purified with flash column chromatography (eluted with CH2Cl2:MeOH = 20:1, the silica gel was neutralized with 1% ammonia solution in DCM prior to using) to give the 5-amino-N-pyrimidinyl-pyrazole triflone 2a (0.014 g, 35% yield) as a white solid.
Mp (DCM): 150–151 °C; 1H NMR (300 MHz, CDCl3) δ: 8.90 (d, J = 4.9 Hz, 2H), 7.48–7.41 (m, 3H), 7.40–7.34 (m, 2H), 6.09 (br s, 2H); 19F NMR (282 MHz, CDCl3) δ: –78.18 (s, 3F).; 13C NMR (126 MHz, CDCl3) δ: 158.96, 157.28, 148.99, 143.03, 130.72, 129.02, 128.75, 128.20, 119.74, 119.68 (q, J = 326.3 Hz), 106.98; IR (KBr): 3411, 3255, 3060, 1600, 1567, 1511, 1428, 1376, 1297, 1213, 1120, 979, 838, 794, 701, 657 cm–1; HRMS (ESI–TOF): calcd for C14H8F3N5O2SCl [M–H]– 402.0039; found 402.0029.
Acknowledgements
This work was supported by JSPS KAKENHI grants JP 16H01142 (Middle Molecular Strategy) and JP16H01017 (Precisely Designed Catalysts with Customized Scaffolding), and ACT-C from the JST (JPMJCR12Z7). ET thanks the Pesticide Science Society of Japan for support.
See Supporting Information (1H, 19F, 13C NMR Spectra) in PDF version
References
- (a) Lee, K. Y.; Kim, J. M.; Kim, J. N. Tetrahedron Lett. 2003, 44, 6737–6740. (b) Elgemeie, G. H.; Zaghary, W. A.; Amin, K. M.; Nasr, T. M. Nucleosides, Nucleotides Nucleic Acids 2005, 24, 1227–1247. (c) McDonald, E.; Jones, K.; Brough, P. A.; Drysdale, M. J.; Workman, P. Curr. Top. Med. Chem. 2006, 6, 1193–2003. (d) Lamberth, C. Heterocycles 2007, 71, 1467–1502. (e) Dolzhenko, A. V.; Dolzhenko, A. V.; Chui, W. K. Heterocycles 2008, 75, 1575–1622. (f) Fustero, S.; Román, R.; Sanz-Cervera, J.-F.; Simón-Fuentes, A.; Cunãt, A. C.; Villanova, S.; Murguía, M. J. Org. Chem. 2008, 73, 3523–3529. (g) Fustero, S.; Sanz-Cervera, J. F.; Simón-Fuentes, A.; Román, R.; Catalán, S.; Murguía, M. ACS Symp. Ser. 2009, 1003, 182–209.
- (a) Kane, J. L.; Hirth, B. H.; Liang, B.; Gourlie, B. B.; Nahill, S.; Barsomian, G. Bioorg. Med. Chem. Lett. 2003, 13, 4463–4466. (b) Kumar, V.; Aggarwal, R.; Tyagi, P.; Singh, S. P. Europ. J. Med. Chem. 2005, 40, 922–927. (c) Aggarwal, R.; Kumar, V.; Tyagi, P.; Singh, S. P. Bioorg. Med. Chem. 2006, 14, 1785–1791.
- (a) Frackenpohl, J.; Gebauer, O.; Cerezo-Galvez, S.; Es-Sayed, M.; Gorgens, U.; Franken, E.-M.; Malsam, O.; Schnatterer, S.; Arnold, C.; Lummen, P.; Schwarz, H.-G.; Hense, S.; Werner, S. PCT Int. Appl. WO 2008077483 A1, 2008. (b) Kudo, N.; Furuta, S.; Taniguchi, M.; Endo, T.; Sato, K. Chem. Pharm. Bull. 1999, 47, 857–868.
- (a) Banks, B. J. Eur. Pat. Appl. EP 846686 A1, 1998. (b) Banks, B. J. U.S. Patent 6,069,157A, 2000.
- Meegalla, S. K.; Doller, D.; Sha, D.-Y.; Soll, R.; Wisnewski, N.; Silver, G. M.; Dhanoa, D. Bioorg. Med. Chem. Lett. 2004, 14, 4949–4953.
- (a) Food Standards Agency: Update on Fipronil in eggs, https://www.food.gov.uk/news-updates/news/2017/16427/update-on-fipronil-in-eggs-10-august; (b) Health assessment of individual measurements of Fipronil levels detected in foods of animal origin in Belgium, DOI 10.17590/20170802-140011
- Schwarz, H.-G.; Schenke, T.; Hense, A.; Velten, R.; Maechling, S.; Werner, S.; Franken, E.-M.; Gorgens, U.; Lummen, P.; E.-Kiintscher, U.; Voerste, A. PCT Int. Appl. WO 2009156090 A2, 2009.
- (a) Filler, R.; Kobayashi, Y. Biomedical Aspects of Fluorine Chemistry; Elsevier Biomedical Press and Kodansha Ltd.: Amsterdam, Tokyo, 1982. (b) Filler, R.; Kobayashi, Y.; Yagupolskii, L. M. (Ed) Organofluorine Compounds in Medicinal Chemistry and Biomedical Applications; Elsevier: Amsterdam, New York, 1993.
- Xu, X.-H.; Shibata, N. J. Synth. Org. Chem. Jpn. 2013, 71, 1195–1201.
- (a) Xu, X.-H.; Taniguchi, M.; Azuma, A.; Liu, G.; Tokunaga, E.; Shibata, N. Org. Lett. 2013, 15, 686–689. (b) Xu, X.-H.; Taniguchi, M.; Wang, X.; Tokunaga, E.; Ozawa, T.; Masuda, H.; Shibata, N. Angew. Chem., Int. Ed. 2013, 52, 12628–12631. (c) Kawai, H.; Sugita, Y.; Tokunaga, E.; Sato, H.; Shiro, M.; Shibata, N. ChemistryOpen 2014, 3, 14–18. (d) Das, P.; Shibata, N. J. Org. Chem. 2017, 82, 11915–11924.
- (a) Shibata, N.; Ishimaru, T.; Nakamura, S.; Toru, T. J. Fluorine Chem. 2007, 128, 469–483. (b) Shibata, N.; Mizuta, S.; Kawai, H. Tetrahedron: Asymmetry 2008, 19, 2633–2644. (c) Shibata, N.; Matsnev, A.; Cahard, D. Beilstein J. Org. Chem. 2010, 6, No. 65. (d) Kawai, H.; Shibata, N. Chem. Rec. 2014, 14, 1024–1040. (e) Shibata, N. Bull. Chem. Soc. Jpn. 2016, 89, 1307–1320.
- (a) Huang, Z.; Okuyama, K.; Wang, C.; Tokunaga, E.; Li, X.; Shibata, N. ChemistryOpen 2016, 5, 188–191. (b) Huang, Z.; Jia, S.; Wang, C.; Tokunaga, E.; Sumii, Y.; Shibata, N. J. Fluorine Chem. 2017, 198, 61–66. (c) Huang, Z.; Wang, C.; Tokunaga, E.; Sumii, Y.; Shibata, N. Org. Lett. 2015, 17, 5610–5613. (d) Das, P.; Gondo, S.; Tokunaga, E.; Sumi, Y.; Shibata, N. Org. Lett. 2018, 20, 558–561.
- (a) Muruganantham, R.; Mobin, S. M.; Namboothiri, I. N. N. Org. Lett. 2007, 9, 1125–1128. (b) Xie, J.-W.; Wang, Z.; Yang, W.-J.; Konga, L.-C.; Xu, D.-C. Org. Biomol. Chem. 2009, 7, 4352–4354. (c) Muruganantham, R.; Namboothiri, I. J. Org. Chem. 2010, 75, 2197–2205.
- Huang, K.; Ma, Q.; Shen, X.; Gong, L.; Meggers, E. Asian J. Org. Chem. 2016, 5, 1198–1203.
Recommended for publication by Prof. Norio Shibata
Fluorine Notes, 2018, 116, 9-10
