Received: сентябрь 2012
Fluorine Notes, 2012, 84, 5-6
Метод синтеза чистых (E)-3-(перфторалкил)-аллиловых спиртов
Peter Ivanko, Csongor Szíjjártó, Dénes Szabó and József Rábai*
Institute of Chemistry, Eotvos Lorand University, P. O. Box 32, H-1518, Budapest 112, Hungary
e-mail: rabai@elte.hu
Аннотация. Предложен простой метод получения (E)-3-(перфторалкил)-аллиловых спиртов (1).
Ключевые слова: (E)-3-(перфторалкил)-аллиловые спирты, 2-иодо-3-перфторалкил-1-пропанолы, дегидрогалогенирование
Перфторалкилпропенолы получают дегидрогалогенированием соответствующих перфтралкил-галогидринов и применяют как прекурсоры для синтеза материалов с низкой поверхтностной энергией. Такие синтезы обычно дают техническую смесь (E)-:(Z) изомеров вместе с побочными продуктами типа 2-перфторалкилметил-оксиранов. Для этих методов применяют неорганические основания (такие как NaOH, KOH, NaHCO3, Na2CO3), алкоксиды, или амидиновые бициклические основания типа DBU (1,8-diazabicyclo[5.4.0]undec-7-ene) или DBN (1,5-Diazabicyclo[4.3.0]non-5-ene) и применяют спиртовые растворители типа (MeOH, EtOH и i-PrOH) при температуре 15 - 100°C и времени синтеза 1-14 часов [1-3].
Перфторалкилпропенолы [(E)-I/(Z)-I] могут быть получены как описано в патенте США в соотношении (E)-:(Z) 93:6 с 69-91% выходом используя N-метилпиррилидон как растворитель и Me4NHCO3 как основание для дегидрогалогенирования начального галогидрина [4].
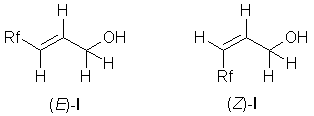
Установлено, что дальнейшее увеличение (E)-:(Z) изомерного соотношения F-алкил-аллиловых спиртов (1a-e) возможно с селективной, но более медленной реакцией дегидроиодирования легко доступных 3-перфторалкил-2-иодо-1-пропанолов (2a-e, “фтор-иодогидрины” [5]) с диэтиламином как основнием в эфире при комнатной температуре. Соединения были получены с высоким выходом и чистотой при времени реакции от 11 до 44 дней. Все попытки увеличить скорость этого типа реакций ужесточением условий реакции приводили к существенному снижению выхода и изомерной чистоты (ср. с определением “Lethargic reaction”, [6]).
Следует отметить, что эти иодогидрины (2a-e) могут быть включены в три различных типа реакций элиминирования HI: преимущественная реакция, приводящая к Rfn-аллиловым спиртам (1a-e), а также реакции, приводящие к фтороксиранам (RfnCH2CHCH2O) или фторальдегидам (RfnCH2CH2CH=O) [7].
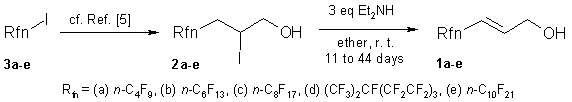
Полученные соединения были охарактеризованы с помощью 1H-, 13C и 19F ЯМР спектров и газовой хроматографии. Хроматографические анализы показали , что содержание транс-изомера составляло 95-97%, в то время как содержание примесей с меньшим временем удерживания не превышало 4%.
Экспериментальная часть
Исходные перфторалкилиодогидрины 2a-e были получены реакцией перфторалкил иодидов 3a-e и алиловых спиртов по методике из работы [5]. 1H-, 13C- и 19F-ЯМР спектры были получены на приборе Bruker Avance 250 при комнастной температуре. Химический сдвиг (δ) представлен в миллионных долях (ppm) относительно внутреннего стандарта TMS (δ=0.00 for 1H, δ=0.00 for 13C) и внешнего стандарта CFCl3 (δ=0.00 for 19F). Для хроматографического анализа применяли хроматограф Hewlett-Packard 5890 Series II, PONA [crosslinked methylsilicone gum] 50 m x 0.2mm x 0.5 mm колонки, газ-носитель H2, детектор ионизации в пламени (FID); хроматографию проводили по следующей программе: 120 °C, 5 мин., 10°C/мин., 250 °C, 5 мин.; Испаритель.: 250°C, Деткетор: 280°C).
Методика синтеза (GP). Раствор иодогидрина (250mmol) в диэтиловом эфире (2mL на 1 г иодогидрина) смешивали с диэтиламином (750mmol) при комнатной температуре. Реакционный сосуд продували аргоном, закрывали и оборачивали алюминиевой фольгой для защиты от солнечного света. Смесь хранили в темноте при комнатной температуре пока не достигалась 99+% конверсия.Твердую соль Et2NH*HI отфильтровывали, промывали эфиром и сушили. После этого фильтрат промывали 5% HCl-H2O и водой и осушали Na2SO4. Продукт выделяли фракционной разгонкой с использованием короткой колонки Vigreaux.
(E)-4,4,5,5,6,6,7,7,7-nonafluoro-hept-2-ene-1-ol (1a). C4F9-Иодогидрин (2a, 100 g, 248 mmol, чистота по ГЖХ 96%) вступал в реакцию согласно методике синтеза в течении 11 дней с образованием 57.05 г (83%) бесцветной маслянистой жидкости, Ткип 85-86 °C/20 mm Hg, чистота по ГЖХ 95.8%. 1H ЯМР (CDCl3) δ: 6.52 (hd, 1H, J= 15.8, 2.2 Hz, RfnCH); 5.95 (qa, 1H, J= 15.6 Hz, CHCH2); 4.32 (m, 2H, J= 3.8, 2.2, 1.6 Hz, CH2OH); 2.93 (s, br, 1H, OH); 13C ЯМР (CDCl3) δ: 61.3 (C1); 116.4 (t, C2, J= 23.4 Hz); 141.6 (t, C3, J= 8.7 Hz); 19F ЯМР (CDCl3) δ: -81.95 (tt, 3F, J= 9.9, 3.0 Hz, CF3); -112.58 (~t, 2F, J= 12.0 Hz, CH2CF2); -125.05 (m, 2F, J= 10.1, 6.9 Hz); -126.59 (m, 2F, J= 6.9, 5.2 Hz).
(E)-4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluoro-non-2-ene-1-ol (1b). C6F13-Иодогидрин (2b, 200 g, 397mmol, чистота по ГЖХ 96%) вступал в реакцию согласно методике синтеза в течении 14 дней с образованием 123 г (82%) бесцветной маслянистой жидкости,Ткип 103 °C/20 mm Hg, чистота по ГЖХf 96.1%. 1H ЯМР (CDCl3) δ: 6.52 (hd, 1H, J= 15.8, 2.2 Hz, RfnCH); 5.95 (qa, 1H, J= 15.0 Hz, CHCH2); 4.33 (m, br, 2H, CH2OH); 2.66 (s, br, 1H, OH); 13C ЯМР (CDCl3) δ: 61.5 (C1); 116.6 (t, C2, J= 23.4 Hz); 141.4 (t, C3, J= 8.5 Hz); 19F ЯМР (CDCl3) δ: -81.77 (tt, 3F, J= 9.9, 2.6 Hz, CF3); -112.43 (tt, 2F, J= 13.4, 3.4 Hz, CH2CF2); -122.46 (br, 2F); -123.73 (br, 2F); -124.17 (m, br, 2F); -127.04 (m, 2F)
(E)-4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoro-undec-2-ene-1-ol (1c). The C8F17-iodohydrin (2c, 200 g, 331mmol, GC assay 97%) was reacted according to the GP for 19 days and worked up to yield 140 g (89%) colorless oil, bp 126-127°C/16 mmHg, with GC purity of 95.5%. 1H NMR (CDCl3) δ: 6.52 (hd, 1H, J= 15.8, 2.2 Hz, RfnCH); 5.96 (qa, 1H, J= 15.2 Hz, CHCH2); 4.33 (m, br, 2H, CH2OH); 2.48 (s, br, 1H, OH); 13C NMR (CDCl3) δ: 61.5 (C1); 116.6 (t, C2, J= 23.4 Hz); 141.4 (t, C3, J= 8.5 Hz); 19F NMR (CDCl3) δ: -81.75 (~t, 3F, J= 9.9 Hz, CF3); -112.40 (~t, 2F, J= 12.9 Hz, CH2CF2); -122.25 (m, br, 2F); -122.75 (m, br, 4F); -123.55 (m, br, 2F); -124.12 (m, br, 2F); -127.00 (m, br, 2F)
(E)-4,4,5,5,6,6,7,7,8,8,9,9,10,11,11,11-hexadecafluoro-10-trifluormethyl-undec-2-ene-1-ol (1d). The iso-C9F19-iodohydrin (2d, 48.3 g, 73.9mmol) was reacted according to the GP for 9 days and worked up to yield 27.7 g (71%) colorless oil, bp 142-144°C/17 mmHg, with GC purity of 94.4%. 1H NMR (CDCl3) δ: 6.52 (hd, 1H, J= 15.8, 2.2 Hz, RfnCH); 5.96 (qa, 1H, J= 14.7 Hz, CHCH2); 4.34 (m, br, 2H, CH2OH); 2.24 (s, br, 1H, OH); 13C NMR (CDCl3) δ: 61.6 (C1); 116.6 (t, C2, J= 23.4 Hz); 141.5 (t, C3, J= 8.7 Hz) 19F NMR (CDCl3) δ: -72.62 (m, 6F, CF3); -112.31 (m, 2F, CH2CF2); -115.70 (s, br, 2F); -121.46 (s, br, 2F); -122.14 (m, br, 4F); -124.02 (m, br, 2F); -186.80 (m, br, 1F).
(E)-4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,12,12,13,13,13-heneicosafluoro-tridec-2-ene-1-ol (1e). The C10F21-iodohydrin (2e, 25.0 g, 41.4mmol) was reacted according to the GP for 44 days and worked up to yield 16.9 g (71%) colorless melt, bp 108-110°C/0.1 mm Hg, mp 54-56 °C/isooctane, with GC purity of 94.4%. 1H NMR (CDCl3) δ: 6.71 (hd, 1H, J= 15.6, 2.4 Hz, RfnCH); 6.06 (qa, 1H, J= 15.2 Hz, CHCH2); 4.33 (s, br, 2H, CH2OH); 2.94 (s, br, 1H, OH); 13C NMR (CDCl3) δ: 61.5 (C1); 115.4 (t, C2, J= 23.4 Hz); 141.5 (t, C3, J= 8.5 Hz); 19F NMR (CDCl3) δ: -81.74 (t, 3F, J= 10.4 Hz, CF3); -112.41 (t, 2F, J= 12.9 Hz, CH2CF2); -122.21 (s, br, 2F); -122.60 (s, br, 8F); -123.51 (s, br, 2F); -124.13 (s, br, 2F); -126.99 (m, br, 2F)
Благодарности. Эта работа была выполнена при поддержке Hungarian Scientific Research Foundation (OTKA K 062191 “Sustainable fluorous chemistry”): Европейский союз и European Social Fund осуществляли поддержку через грант no. TÁMOP_4.2.1./B-09/1/KMR-2010-0003.
Список литературы
- GB1101049, Daikin Kogyo Kabushiki Kaisha, Patent filed: May 19, 1966.
- JP 52144610, A. Yoshio, D. Shigeo, K. Yasuhisa, O. Kazuo, (Daikin Kogyo), 1977.
- US4278552, I. Hisamoto, et al. (Daikin Kogyo, Co.,Ltd) Patent filed: April 12, 1979. (et al. = N. Enjo, C. Maeda, T. Esaka, Y. Omure, T. Iida, H. Aomi)
- US Patent 4754082, K. Raab, J. J. Pöschl, (Hoechst), Patent filed: July 2, 1987.
- J. Rábai J, Cs. Szíjjártó, P. Ivanko, D. Szabó. Synthesis 2007, 2581-2584.
- D. E. Pearson, O. D. Keaton, J. Org. Chem. 1963, 28, 1557-1958.
- P. Ivanko, F. T. Takács, J. Rábai, Convergent synthesis of fluorous reagents. 14th European Symposium on Fluorine Chemistry, Poznań, Poland, 2004. Book of Abstracts, p. 84
Материал рекомендован к публикации членом редколлегии С.М. Игумновым
Fluorine Notes, 2012, 84, 5-6
