Received: November 2016
DOI 10.17677/fn20714807.2016.06.01
Fluorine Notes, 2016, 109, 1-2
Study of Reactivity of Trifluoromethanesulfonyl Bromide Towards Unsaturated Hydrocarbons
A.A. Tyutyunovab, L.F. Ibragimovaa, N.D. Kagramanova, S.R. Sterlina, S.M. Igumnovab
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, ul. Vavilova 28, V-334, GSP-1, Moscow, 119991 Russia
bNPO PiM-INVEST LLC, ul. Vavilova 28, Moscow, 119991 Russia
e-mail: tuytuynov@rambler.ru
Abstract: The interaction between trifluoromethanesulfonyl bromide and unsaturated hydrocarbons is studied under the conditions of photochemical, thermochemical, and ion-radical initiation.
Keywords: Trifluoromethanesulfonyl bromide, trifluoromethylation.
Aliphatic compounds containing a terminal trifluoromethyl group with the overall formula of CF3(CH2)nX (I), where n = 2-10, X = OH, Br, I, NH2, SH, CHO etc. are marketable intermediate organic chemicals and can be used for preparing pharmaceuticals [1-6], agricultural chemicals [7-8], surfactants [9], liquid crystals [10] etc. Besides, carbinols I (X = OH, n = 2-4) are of independent interest in practice and can be used as components of lubricants [11], cleaning composite materials [12], eluents [13], and extractants [14]. These alcohols are characterized by a short half-life in the atmosphere and low values of the global warming potential (GWP), which in some cases allows using them as an alternative to freons [15].
The main methods of synthesis of linear ω-trifluoromethyl-containing aliphatic derivatives are both addition of polyhaloalkanes (CCl4, CCl3Br) to terminal olefins [16] with the further fluorination of the adducts [8, 17] and direct addition of the CF3-group to the hydrocarbon chain using reagent presented below (scheme 1, table 1) [18-26].
Scheme 1
Table 1. Trifluoromethyl Containing Reagents and Conditions of Their Reactions with Unsaturated Compounds.
CF3-Reagent |
Conditions |
CF3I Tbp = -22°C |
200°C [27]; 80°C, AIBN [28]; UV-light [27, 29]; Na2S2O4, NaHCO3 [30]; MLn catalyst [20]; Photoredox catalyst [21] |
CF3Br Tbp = -59°C |
Zn, CuI, Ultrasound [31]; Zn, Cp2TiCl2, Ultrasound [31]; 200°C, Pd(PPh3)4 [32]; Aluminum chlorofluoride, 50°C [33] |
CF3SO2Cl Tbp = 32°C |
90÷145°C, ROOR [34]; 125°C, Ru(PPh3)3Cl2 [35-36]; UV-light [24]; Photoredox catalyst [37-41] |
CF3SO2Na |
tBuOOH, Cu salt [42-44]; AgNO3, K2S2O8 [45]; Photoredox catalyst [46-47] |
CF3SO2Br Tbp = 60°C |
20÷65°C [48-49]; sunlight [shown for RFSO2Br [50-51]; 55÷95°C, ROOR [48, 51-52] |
CF3SiMe3 Tbp = 55°C |
CuTc, PhI(OAc)2, K2CO3 [53]; AgNO3, PhI(OAc)2, AcONa [54] |
|
MLn catalyst [22, 24] |
|
TMSN3, [Cu(CH3CN)4]PF6 [55]; CuBr2, SOX2 [56]; MLn catalyst [22, 26, 57] |
CF3CCl3 Tbp = 46°C |
Fe, P(OEt)3, 120°C [58]; UV-light [59] |
CF3CH2I Tbp = 55°C |
Na2S2O4, NaHCO3 [60]; iPr2NEt, fac-Ir(ppy)3, visible light [61] |
CF3CH2SO2Cl Tbp = 140°C |
K2HPO4, fac-Ir(ppy)3, visible light [62]; |
Trifluoromethanesulfonyl bromide CF3SO2Br (1) is of special interest among the chemicals used for direct trifluoromethylation of unsaturated compounds (table 1) as the most convenient trifluoromethylating agent from a synthetic viewpoint. At the same time, the examples of application of compound 1 in synthesis of fluoro-containing compounds in the literature are fragmentary [24, 48-49, 52], which led us to performing a systematic investigation of trifluoromethylation of unsaturated hydrocarbons using sulfonyl bromide 1 obtained by smooth bromination of potassium trifluoromethanesulfinate synthesized by in situ sulfodioxidation of (trifluoromethyl)trimethylsilane (2) [63] or commercially available sodium trifluoromethanesulfinate (3) [48].
Scheme 2
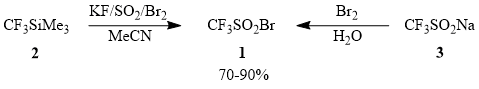
In work with trifluoromethanesulfonyl bromide (1), one must take into account that this compound can be distilled without decomposition (b.p. 58÷60°C), can be kept in dark for a long time, is very photosensitive, and undergoes desulfodioxidation with formation of CF3Br under the action of sunlight. A similar process is observed when compound 1 is heated above 100°C.
Scheme 3
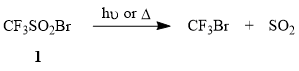
It was shown earlier that compound 1 is added to electron-donating olefins, e.g., aliphatic terminal alkenes, cyclohexene, allyl acetate at 20÷65°C and the reaction is accompanied by elimination of SO2 [48-49]. However, despite the simplicity of the method, one must take into account that the scaling of radical processes may result in the reaction getting out of hand and ending in an explosion (as, e.g., in the case of addition of CCl4 to ethylene catalyzed by benzoyl peroxide [64]).
We found that addition of CF3SO2Br (1) to allyl trifluoroacetamide (4a) and allyl acetate (4b) in sealed tubes under sunlight irradiation (for samples of compounds 4a-b of 2-1 mol) occurs smoothly and results in formation of adducts 5a-b in 70-90% yield, while addition of compound 1 to homoallyl acetate (4c) under similar conditions could be achieved only at the loading of 3-4 mmol of compound 4c. Experiments on a larger scale, with 200-250 mmol of compound 4c inevitably ended in explosion of the tube.
Scheme 4
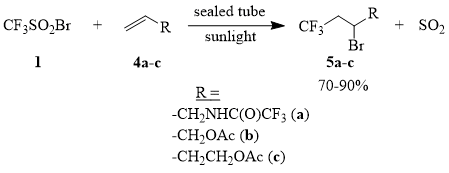
Addition of compound 1 to compounds 4a-b under the conditions of thermal initiation is carried out at the temperatures of 70÷100°C. Thus, in the case of the heating of compounds 1 and 4a (at the molar ratio of 1:1; 100°C/2.5 h), the yield of compound 5a is approximately 50%. One must point out that the reaction between compounds 1 and 4b in hexane (at the molar ratio of 1:1; 65°C/2 h) in an open system results in formation of compound 5b in only 20% yield, which is much less than the yield of compound 5b according to the lit. data [48].
In view of the fact that the rate of desulfooxidation of compound 1 increases significantly under the conditions of the reaction, which results in a decrease in the yield of the target products, thermally initiated addition of sulfonyl bromide 1 to olefins 4a-b in an open system is hardly of any practical interest.
It is known that initiation of radical addition of polyhaloalkanes to olefins can be efficiently carried out using diverse redox systems, e.g., those based on salts of metals of variable valency, such as Cu, Fe, Mn, Ce etc. [65]. It has been shown earlier that copper halides are the most active catalysts of addition of sulfonyl chlorides and sulfonyl bromides to a number of olefins and acetylenes [66-68]. Indeed, it turns out that metallic copper efficiently initiates the addition of compound 1 to compounds 4a-d in an acetonitrile solution already at the room temperature. This process can be smoothly scaled and can be used for synthesis of trifluoromethylated compounds 5a-d.
Scheme 5
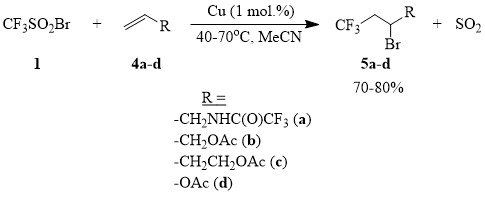
Compounds 5a-d are valuable intermediates that can be used for synthesis of a variety of ω-trifluoromethyl-containing linear aliphatic derivatives [69].
Scheme 6
It is known that radical addition to internal olefins is hindered as compared to similar reactions with terminal olefins, which is related to the steric shielding of the vinyl carbon atom [70-71]. It is no wonder that addition of sulfonyl bromide 1 to butene 14 (sealed tube, insolation) occurs rather slowly and is accompanied by side reactions: formation of CF3Br as a result of desulfodioxidation of compound 1 and trifluoromethanesulfinic acid due to hydrogen abstraction from allylic position by the CF3SO2-radical (scheme 7, table 2). The same reaction in the MeCN medium induced by powdered copper led to formation of adduct 15 in preparative yield, which allowed using it as the starting compound for synthesis of trifluoroisoprene 17 (scheme 8).
Scheme 7
Table 2. Reaction of CF3SO2Br (1) with 1,4-diacetoxy-2-butene (14)a.
Conditions |
Ratio of reaction products, % |
||
15 |
CF3Br |
CF3SO2H |
|
sunlight, 3 daysb |
51 |
34 |
7 |
Cu (2 mol.%), MeCN, rt, 2 days |
81c |
8 |
10 |
a The ratio of compound 1 : compound 14 is 1:1.
b A sealed tube, the conversion of compound 1 is 93%; the loading is 1 mmol.
c The preparative yield is 70% (a mixture of stereoisomers).
Scheme 8
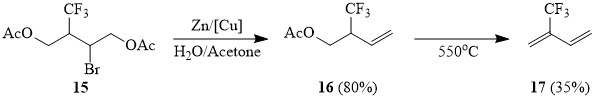
Olefins with an electron-deficient double bond, methyl acrylate (18a) and acrylonitril (18b), also react with sulfonyl bromide 1 in the presence of copper in acetonitrile, however, as expected, the yields of adducts 19a-b are lower, as compared with the above examples. Here, one must point out that telomer-homologs 18a-b are not formed. Under the conditions of photochemical initiation, compound 1 reacts with methyl acrylate forming adduct 19a with a low yield, but formation of telomer-homologs 19a is observed in this case (cf. [51]). Under similar conditions, we failed to involve olefin 18b in the reaction.
Scheme 9
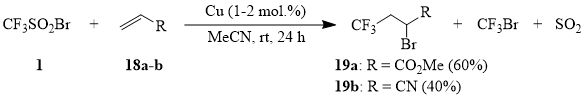
Unexpectedly, it turned out that acrolein (20) reacts with compound 1 with formation of sulfone 21; besides, 2-bromo-4,4,4-trifluorobutanal, the expected product of addition of the CF3-radical, has not been detected among the reaction products.
Scheme 10
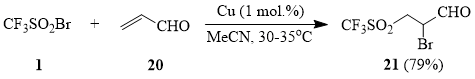
The reaction of compound 1 with styrene (22) resulted in formation of sulfone 24 as the main reaction product that has never been reported before [48].
Scheme 11
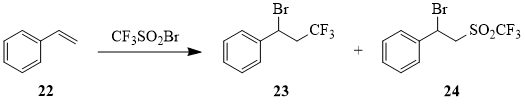
Table 3. Reaction of CF3SO2Br (1) with styrene (22)a.
Conditions |
Ratio of reaction products, % |
|
23 |
24 |
|
90°C, Ph(COO)2, hexane |
64b |
- |
sunlight, hexane |
34 |
65 |
Cu (1 mol.%), MeCN, 25–30°C |
25 |
75c |
a The ratio of compound 1 : compound 22 is 1:1.
b According to the data of [48].
c When reaction products were extracted by vacuum distillation, sulfone 24 undergoes dehydrobromination (by 50%) with formation of PhCH=CHSO2CF3 (25).
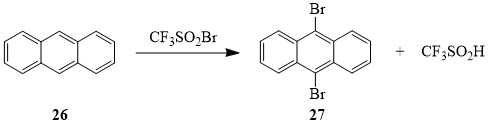
The products of the reaction between sulfonyl bromide 1 and anthracene (26) – 9,10-dibromoanthracene (27) and trifluoromethanesulfinic acid – indicate that in this case compound 1 acts solely as a brominating agent; no products of substitutive or additive trifluoromethylation of anthracene were found.
Scheme 12
Thus, it is shown that trifluoromethanesulfonyl bromide (1) interreacting with reagents of different nature can be a source of trifluoromethyl or trifluoromethanesulfonyl group or act as a brominating agent. At the same time, it is found that compound 1 can enter additive reactions with a sufficiently wide spectrum of unsaturated hydrocarbons under the conditions of photochemical and ion-radical catalysis, which is a convenient preparative method of synthesis of different classes of organic compounds containing trifluoromethyl group.
Experimental
1H, 19F NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 300 and 282 MHz, accordingly; the external standard was CDCl3. Chemical shifts for 1H spectra are presented vs. the residual signal of the solvent (δ 7.26) and are given in ppm vs. tetramethylsilane. Chemical shifts in 19F spectra are given in ppm vs. CFCl3. Downfield shifts are positive. Mass spectra are recorded using a Finnigan Polaris Q mass spectrometer (Trace GC ultra).
Trifluoromethanesulfonyl bromide (1).
A: Synthesis from CF3SiMe3 (2).
The process is potentially dangerous and can be accompanied by heating and ejection of the reaction mixture.
The acetonitrile (2.5 l containing 0.04-0.05% H2O) and 280 g (4.82 mol) of dry KF are successively loaded into a 6 l flask equipped with mechanical stirrer, thermometer, dropping funnel, and a reflux condenser connected to a Tishchenko flask with H2SO4(conc.). Then the solution of 720 g (5.06 mol) of (trifluoromethyl)trimethylsilane (2) and 340 g (5.3 mol) of SO2 in 1 l of acetonitrile is added dropwise under stirring keeping the temperature at 15÷20°C.
The reaction mixture is stirred at 15÷20°C/3-6 h (the reaction mixture heating is possible; the temperature must be controlled by external cooling). In the course of the reaction, the reaction mixture becomes light-brown. If the reaction mixture remains practically colorless during this time, the stirring is ceased and the reaction mixture is left overnight. Then the reaction mixture stirring is renewed and its temperature is maintained in the range of 20÷30°C. After the end of the reaction, the reaction mixture becomes light-brown.
Then the reaction mixture is slowly heated to the temperature of 80÷95°C and low-boiling fraction is distilled off. Distillation is carried out until the vapour temperature of 80°C is reached.
The reaction mixture is cooled to 0÷5°C and 779 g (4.87 mol) of bromine are added dropwise. Then the mixture is stirred for 15-30 min, poured into a 1.5-fold volume of ice water; the lower layer is separated, washed four times with an equal volume of ice water, P2O5 (30 g) is added to organic layer and distilled.
The compound 1 (720 g, 70%) is obtained with the purity of 85-90%; the impurities are FSiMe3 and acetonitrile, b.p. 55÷65°C. 19F NMR δ: -78 (s, CF3).
B: Synthesis from CF3SO2Na (3) [48].
The commercial sodium trifluoromethanesulfinate (3) [1100 g (4.58 mol), the purity ~ 65 wt.%; the impurities are NaBr, Na2SO4, Na3PO4] is added to 3 l of water under stirring. The solution obtained is stirred for 10-15 min and bromine (740 g, 4.63 mol) is added dropwise at 5÷10°C until the stable bromine colouring of the reaction mixture appears.
The lower layer is separated, washed with 1.5-fold volume of ice water; 30 g of P2O5 are added under stirring and the whole is distilled.
The trifluoromethanesulfonyl bromide (1) (878 g, 90%) is obtained with a negligible amount of a bromine admixture that can be removed by addition of 0.5-1 wt.% of decene-1 and further distillation. The purity of compound 1 thus obtained is 99%, b.p. 58÷60°C.
N-(2-bromo-4,4,4-trifluorobutyl)-2,2,2-trifluoroacetamide (5a).
The reactions in sealed tubes are potentially dangerous and can be accompanied by an explosion.
A mixture of 590 g (2.77 mol) of trifluoromethanesulfonyl bromide (1) and 424 g (2.77 mol) of allyltrifluoroacetamide (4a) is sealed into a 1 l glass tube made of molybdenum glass (the diameter 50 mm, the length 500 mm) and exposed to sunlight for 1-4 days (depending of the sunlight intensity) on the window sill. The tube is cooled (-20°C) and opened; boiling stones are added and the whole is slowly heated to 25÷30°C at a rate, at which the boiling of SO2 is not too vigorous. The product obtained is purified by rectification at 0.5-1 torr; rectification in vacuum of 15 torr causes an increase in the product loss due to its partial degradation.
The compound 5a (586-670 g, 70-80%) with the purity of 96% was obtained, b.p. 130°C/15 torr.
1H NMR δ: 2.7 (m, 2H, CF3CH2), 3.7 (m, 2H, CH2NH), 4.2 (m, 1H, CHBr), 8 (br.s, 1H, NH); 19F NMR δ: -78 (s, 3F, C(O)CF3), -66 (t, 3F, 3JHF = 8 Hz, CF3CH2).
2-Bromo-4,4,4-trifluorobutyl acetate (5b).
A mixture of 42.6 g (0.2 mol) of trifluoromethanesulfonyl bromide (1) and 20 g (0.2 mol) of allylacetate (4b) is sealed into a 50 ml glass tube made of molybdenum glass (the diameter 23 mm, thе length 135 mm) and exposed to sunlight for 1-3 days (depending of the sunlight intensity) on the window sill. The tube is cooled (-20°C) and opened; boiling stones are added and the whole is slowly heated to 25÷30°C, while SO2 is removed at a moderate rate. The obtained product is purified by rectification in vacuum of 10 torr.
The compound 5b (39.8 g, 80%) with the purity of 98% is obtained, b.p. 73÷76°C/10 torr.
1H NMR δ: 2 (s, 3H, CH3), 2.6-2.9 (br.m., 2H, CF3CH2), 4.2 (m, 3H, CH2O+CHBr); 19F NMR δ: -65 (t, 3F, 3JHF = 8 Hz, CF3).
Reactions of compound 1 with alkenes 4a-d in the presence of copper (general procedure).
The powdered copper (1.27 g, 0.02 g-at) is added to the solution of olefin 4a-d (2 mol) in 400 ml of acetonitrile, then trifluoromethanesulfonyl bromide (1) (426 g, 2 mol) is added dropwise under stirring at 65÷70°C in the case of 4b, c and 40÷45°C in the case of 4a, d, evolution of SO2 starts after a half-volume of compound 1 is added. The mixture is stirred for 15-60 min when heated, the solvent is evaporated and the residue is distilled at 10-0.5 torr to give target compounds 5a-d.
3-Bromo-5,5,5-trifluoropentyl acetate (5c).
The yield is 80%, b.p. 80÷84°C/0.5 torr.
1H NMR δ: 1.9 (s, 3H, CH3), 2.0-2.3 (br.m, 2H, CH2CH2O), 2.8 (m, 2H, CF3CH2), 4.1 (m, 1H, CHBr), 4.2 (m, 2H, CH2O); 19F NMR δ: -65 (t, 3F, 3JHF = 8 Hz, CF3).
1-Bromo-3,3,3-trifluoropropyl acetate (5d).
The yield is 70%, b.p. 30°C/10 torr.
1H NMR δ: 2.3 (s, 3H, CH3), 3.2-3.5 (br.m., 2H, CH2), 7 (m, 1H, CHBr); 19F NMR δ: -65 (t, 3F, 3JHF = 8 Hz, CF3). The mass spectrum (M/Z, reference): 235[M+H]+, 191[C3H3BrF3O]+, 175[M-CH3CO2]+, 165[M-CF3]+, 155[C3H2BrF2]+, 129[CBrF2]+, 111[CHBrF]+, 95[C3H2F3]+, 91[C4HF2O]+, 75[C3HF2]+ , 64[C2H2F2]+, 51[CHF2]+, 43[C2H3O]+(100%), 36[C3]+.
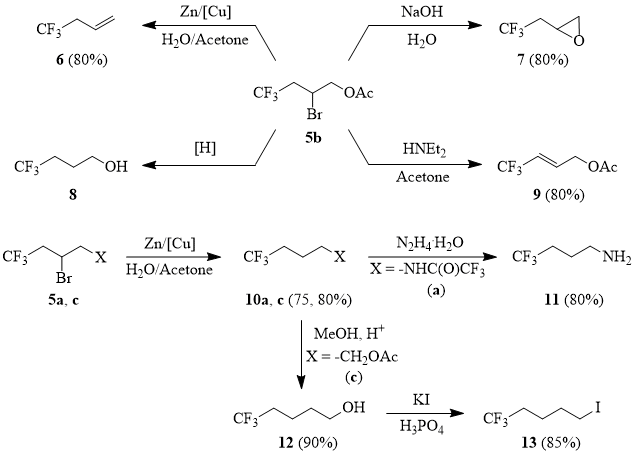
4,4,4-Trifluorobutyl-1-ene (6).
The powdered cuprous chloride (0.5 g 2.5 mmol) is added to the suspension of 6.28 g (0.096 g-at) of powdered zinc in 100 ml of water under stirring at 20°C and the mixture is stirred for 10-15 min. Then a solution of 20 g (0.08 mol) of 2-bromo-4,4,4-trifluorobutyl acetate (5b) in 10 ml of acetone is added dropwise under stirring at 5°C; the reaction mixture is stirred for 1 h at 20°C, heated to the temperature of 50÷80°C; the reaction product is removed into a cooled receiver (0°C). The further rectification gave 7 g (80%) of compound 6, b.p. 10÷12°C.
1H NMR δ: 3.7 (m, 2H, CF3CH2), 6.1 (m, 2H, CH=CH2), 6.6 (m, 1H, CH=CH2); 19F NMR δ: -68 (t, 3F, 3JHF = 14 Hz, CF3).
2-(2,2,2-Trifluoroethyl)oxirane (7).
A mixture of 20 g (0.08 mol) of 2-bromo-4,4,4-trifluorobutyl acetate (5b), 7.7 g (0.24 mol) of methanol and 0.1 ml of 50% sulfuric acid is refluxed with Vigreux column, while methyl acetate and methanol are slowly distilled using off. Then the Vigreux column is removed and the residues of methanol are distilled from the reaction mixture. Then the solution of 3.85 g (0.096 mol) of sodium hydroxide in 5.8 ml of water is added dropwise to the reaction mixture under stirring at 100°C with simultaneous distillation of product. Further rectification gave 8 g 7, b.p. 72÷75°C.
1H NMR δ: 2.5-2.9 (br.m, 2H, CF3CH2), 2.6-2.9 (br.m, 2H, CF3CH2), 2.9, 3.2, 3.5 (m, 1H+1H+1H, an oxirane ring); 19F NMR δ: -66 (t, 3F, 3JHF = 11 Hz, CF3).
(2E)-4,4,4-Trifluorobutyl-2-ene-1-ylacetate (9).
A mixture of 58 g (0.23 mol) of 2-bromo-4,4,4-trifluorobutyl acetate (5b) and 33.6 g (0.46 mol) of diethylamine in 200 ml of acetone is refluxed for 4-5 h. The mixture is cooled, diluted with triple volume of water; the product is extracted with 100 ml of CH2Cl2; the obtained solution is washed with cold water; the solvent is evaporated. The rectification of residue gave 31 g (80%) of compound 9, b.p. 135÷138°C. The ratio of cis- and trans-isomers is 1:9.
1H NMR δ: 2.5 (s, 3H, CH3), 5.2 (br.s, 2H, CH2), 6.4 (m, 1H, CH=), 7 (m, 1H, CH=); 19F NMR δ: -65 (s, 3F, CF3). The mass spectrum (M/Z, reference): 169[M+H]+, 148[M-HF]+, 128[M-2HF]+, 109[M-CO2Me]+, 106[M-Ac-F]+, 95[C3H2F3]+, 89[M-AcO-HF]+, 77[C3H3F2]+, 69[CF3]+, 59[AcO]+, 51[CF2H]+, 43[C2F]+(100%), 39[C3H3]+.
2,2,2-Trifluoro-N-(4,4,4-trifluorobutyl)acetamide (10a).
The mixture of powdered zinc (260 g, 3.97 mol), cuprous chloride (19 g, 0.095 mol), 2 l of water and 0.3 l of acetone is stirred in a 6 l flask equipped with mechanical stirrer, thermometer, dropping funnel and a reflux condenser for 15-20 min, cooled to 5°C, and the solution of 1000 g (3.31 mol) of N-(2-bromo-4,4,4-trifluorobutyl)trifluoroacetamide (5a) in 0.4 l of acetone is added at such a rate that the mixture temperature does not rise above 10°C. Then external cooling is removed; the reaction mixture is stirred for 5-6 h; here, the mixture temperature rises to 30°C; then the mixture is left overnight.
Then the reaction mixture is cooled to 15°C and 0.6 l of concentrated hydrochloric acid are slowly added under stirring, while avoiding vigorous hydrogen evolution. The mixture temperature is warmed up to 20°C; the stirring is stopped; the reaction mixture is layered; then the lower layer is separated, filtered through a paper filter, and washed with a 1.5-fold volume of 5% hydrochloric acid. The product obtained contains acetone and 4,4,4-trifluorobutyl-1-ene (6) as impurities that are removed by distillation when the mixture is heated to 80÷100°C. The residue contains 656 g (80%) of compound 10a (purity 90%) that was used hereafter without additional purification. The analytical sample was isolated by rectification, b.p. 110÷115°C/10 torr; crystals, m.p. 36°C.
1H NMR δ: 2.3 (m, 2H, CF3CH2CH2), 2.6 (m, 2H, CF3CH2), 3.8 (m, 2H, CH2NH), 9.1 (br.s, 1H, NH); 19F NMR δ: -78 (s, 3F, C(O)CF3), -68 (t, 3F, 3JHF = 11 Hz, CF3CH2).
4,4,4-Trifluorobutylamine (11).
A mixture of 656 g (2.65 mol) of amide 10a (with the purity of 90% obtained according to the procedure described above) and 160 g (3.2 mol) of hydrazine hydrate is heated with distillation of the product boiling at 80÷105°C. The distillate obtained is washed with an equal volume of 30% aqueous solution of NaOH and distilled over solid KOH to give 268 g ( 80%) of compound 11 (purity 98%), b.p. 86÷87°C.
1H NMR δ: 1.5 (m, 2H, CF3CH2CH2), 2.1 (m, 2H, CF3CH2), 2.6 (t, 2H, CH2NH2), 3.3 (br.s, 2H, NH2); 19F NMR δ: -68 (t, 3F, 3JFH = 8 Hz, CF3).
5,5,5-Trifluoroamyl acetate (10c).
Cuprous chloride (3.96 g, 0.02 mol) is added under stirring to the suspension of 59.6 g (0.91 g-at) of powdered zing in 400 ml of water. The mixture is stirred for 15-20 min, cooled to the temperature of 5°C, and the solution of 200 g (0.76 mol) of 3-bromo-5,5,5-trifluoropentyl acetate (5c) in 140 ml of acetone is added at such a rate that the mixture temperature does not rise above 10°C. Then the external cooling is removed; the reaction mixture is stirred for 4-5 h and left overnight.
Then the reaction mixture is cooled to 15°C and 130 ml of concentrated hydrochloric acid is added dropwise under stirring. The reaction mixture is warmed up to 20°C, the stirring is ceased, the reaction mixture is layered and the lower layer is separated, filtered through a paper filter, and distilled in vacuum to give 112 g (80%) of compound 10c that was used hereafter without additional purification.
1H NMR δ: 2.0 (m, 4H, CH2CH2CH2O), 2.3 (s, 3H, CH3), 2.5 (m, 2H, CF3CH2), 4.4 (t, 2H, CH2O); 19F NMR δ: -68 (t, 3F, 3JHF = 11 Hz, CF3).
5,5,5-Trifluoropentanol (12).
A mixture of 100 g (0.54 mol) of 5,5,5-trifluoropentyl acetate (10c), 52 g (1.62 mol) of methanol, and 0.5 ml of 50% sulfuric acid is refluxed with Vigreux column, while methyl acetate and methanol are slowly distilled off, the residue is distilled to give 69 g (90%) of compound 12, b.p. 150°C.
1H NMR δ: 1.8 (m, 4H, CH2CH2CH2O), 2.3 (m, 2H, CF3CH2), 3.8 (t, 2H, CH2OH), 5.3 (s, 1H, CH2OH); 19F NMR δ: -68 (t, 3F, 3JHF = 8 Hz, CF3).
1,1,1-Trifluoro-5-iodopentane (13).
Phosphorus pentoxide (400 g, 1.41 mol) is added portion-wise under stirring to 600 g (5.2 mol) of 85% orthophosphoric acid so that the mixture temperature does not exceed 100°C. The mixture is stirred for 40 min, cooled to the room temperature and 459 g (2.77 mol) of KI and 200 g (1.41 mol) of 5,5,5-trifluoropentanol (12) are added. The reaction mixture is stirred at 105÷110°C for 5-6 h and left overnight.
Cold water (1 l) and methylene chloride (0.2 l) are added to the reaction mixture. The mixture is stirred for 10 min and then the lower layer is separated and washed with an equal volume of cold water and the Na2SO3 solution until the discoloration of the organic layer. The solvent is evaporated and the residue is distilled at reduced pressure (10 torr), collecting the fraction that boils within the range 55÷60°C. Further rectification gave 302 g (85%) of compound 13 (purity 98%).
1H NMR δ: 1.4, 1.6 (m, 2H+2H, CH2CH2CH2I), 1.8 (m, 2H, CF3CH2), 2.9 (t, 2H, CH2I); 19F NMR δ: -68 (t, 3F, 3JHF = 10 Hz, CF3).
4-(Acetoxy)-2-bromo-3-(trifluoromethyl)butyl acetate (15).
Powdered copper (0.63 g, 0.01 g-at) is added to the solution of 100 g (0.58 mol) of (2E)-1,4-diacetoxy-2-butene (14) in 100 ml of acetonitrile. The solution is bubbled with argon and then trifluoromethanesulfonyl bromide (1) (123.5 g, 0.58 mol) is added under stirring. The reaction mixture is stirred for two days at the room temperature; the solvent is evaporated and the residue is distilled at a pressure of 0.5 torr. The further rectification of the distillate obtained gave 130 g (70%) of adduct 15 (a mixture of stereoisomers), b.p. 95÷105°C/0.5 torr.
1H NMR δ: 2.2 (two s, 6H, C(O)CH3), 3.4-3.5 (m, 2H, CHBr+CF3CH), 4.6 (d, 4H, OCH2); 19F NMR δ: -68 (d, 3JHF = 8 Hz, CF3), -65 (d, 3JHF = 8 Hz, CF3) with the ratio of 1:1.
2-(Trifluoromethyl)butyl-3-enyl acetate (16).
Compound 10c is obtained in a similar way in 80% yield, b.p. 45÷55°C/10 torr.
1H NMR δ: 2.5 (s, 3H, C(O)CH3), 3.7 (m, 1H, CF3CH), 4.7 (m, 2H, OCH2), 5.9 (m, 2H, CH=CH2), 6.2 (m, 1H, CH=CH2); 19F NMR δ: -70 (d, 3F, 3JHF = 8 Hz, CF3).
2-(Trifluoromethyl)butyl-1,3-diene (17).
2-(Trifluoromethyl)butene-3-yl acetate (16) is passed dropwise in the flow of argon through a Pyrex tube with glass filling at 540÷550°C. The reaction product is collected in a cooled receiver (-10÷-20°C) connected to the outlet of the reaction tube. Compound 17 is obtained by distillation of condensate in 35% yield (purity ~ 90%), b.p. 35÷45°C.
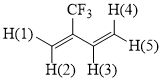
1H NMR δ: 5.9 (d, 1H, H(5)), 6.2 (s, 1H, H(1)), 6.25 (d, 1H, H(4)), 6.35 (s, 1H, H(2)), 6.95 (dd, 1H, H(3)); 19F NMR δ: -68 (s, 3F, CF3). The mass spectrum (M/Z, reference): 122[M]+(100%), 121[M-H]+, 103[M-F]+, 102[M-HF]+, 101[M-HF-H]+, 83[CH2=C(CF)CH=CH]+, 77[C3H3F2]+, 75[CF2CCH]+, 69[CF3]+, 57[C3H2F]+, 53[M-CF3]+, 51[CF2H]+.
Methyl-2-bromo-4,4,4-trifluorobutanoate (19a).
The powdered copper (0.15 g, 2.4 mg-at) is added to the solution of 20 g (0.23 mol) of freshly distilled methyl acrylate (18a) in 30 ml of acetonitrile. The solution is bubbled with argon and then 49 g (0.23 mol) of trifluoromethanesulfonyl bromide (1) are added under stirring. The reaction mixture is stirred for two days at the room temperature, the volatile products are distilled off into a cooled trap (-50°C) at 10 torr and the residue is distilled at the same pressure. Further rectification gave 32 g (60%) of compound 19a, b.p. 50÷55°C/10 torr.
1H NMR δ: 2.85 (m, 1H, CF3CHA), 3.2 (m, 1H, CF3CHB), 3.8 (s, 3H, OCH3), 4.5 (t, 1H, CHBr); 19F NMR δ: -66 (t, 3F, 3JHF = 10 Hz, CF3). The mass spectrum (M/Z, reference): 235[M+H]+, 203[M-OMe]+, 175[M-CO2Me]+, 174[M-CO2Me-H]+, 155[M-CO2Me-HF]+, 135[M-Br-HF]+, 111[CHBrF]+, 95[M-CO2Me-Br-H]+, 91[CF2=CH-CH-CH3]+(100%), 77[M-CO2Me-F-Br]+, 69[CF3]+, 59[CO2Me]+, 51[CF2H]+.
2-Bromo-4,4,4-trifluorobutyronitrile (19b).
Compound 19a is obtained in a similar way in 40% yield, b.p. 53÷57°C/10 torr.
1H NMR δ: 2.85-3.15 (m, 2H, CF3CH2), 4.65 (t, 1H, CHBr); 19F NMR δ: -66 (t, 3F, 3JHF = 9 Hz, CF3). The mass spectrum (M/Z, reference): 201[M]+, 182[M-F]+, 163[M-F-HF]+, 155[M-F-CN-H]+, 146[M-3F-H]+, 132[M-CF3]+, 129[M-CF3-3H]+, 122[M-Br]+(100%), 106[M-CF3-CN]+, 102[M-Br-HF]+, 94[M-Br-CN-2H]+, 75[M-Br-CN-H2-F]+, 69[CF3]+, 52[C2H2CN]+, 51[CF2H]+.
2-Bromo-3-(trifluoromethanesulfonyl)propanal (21).
Trifluoromethanesulfonyl bromide (1) (57 g, 0.268 mol) is added dropwise to the solution of 15 g (0.268 mol) of freshly distilled acrolein (20) in 20 ml of acetonitrile containing 0.1 g (1.6 mg-at) of powdered copper under stirring at 25÷35°C. The mixture is stirred for 3 h; the volatile products are distilled off into a cooled trap (-50°C) under reduced pressure (10 torr). The distillation of the residue afforded 57 g (the yield of 79%) of the fraction that boils within the range 80÷95°C/1 torr. Further rectification gave analytical sample of compound 21, b.p. 80÷85°C/1 torr.
1H NMR δ: 3.85, 4.25 (ABq, 2H, 2JHH = 30 Hz, CH2), 4.9 (t, 1H, CHBr), 9.4 (s, 1H, CHO); 19F NMR δ: -80 (s, 3F, CF3SO2). The mass spectrum (M/Z, reference): 269[M+H]+, 203[C4H3BrF3O]+, 189[M-Br]+, 135[C4H4BrO]+, 133[CF3SO2]+, 119[C4HF2S]+, 109[CHBrO]+, 108[CBrO]+, 107[C2H3Br]+, 106[C2H2Br]+(100%), 82[CF2S]+, 75[C3H4FO]+, 69[CF3]+, 65[C2H3F2]+, 55[C3H3O]+, 48[SO]+, 39[C3H3]+.
Reaction of compound 1 with styrene 22.
Synthesis of (1-bromo-3,3,3-trifluoropropyl)benzene (23) and 1-bromo-2-(trifluoromethanesulfonyl)ethylbenzene (24).
A: Reaction under sunlight irradiation.
The solution of 2 g (9.4 mmol) of trifluoromethanesulfonyl bromide (1) and 1 g of (9.6 mmol) of freshly distilled styrene (22) in 6 ml of hexane is prepared. Approximately 0.3 ml of this solution is sealed in a molybdenum glass tube (the diameter 3 mm, the length 100 mm) and exposed to sunlight for 1 day on the window sill. Then the tune is placed into a standard 5 mm NMR tube, CDCl3 is added as an external standard and the reaction mixture is analyzed using the 19F NMR technique.
According to the data of 19F NMR, the mixture contains 34% of compound 23 (δ: -65, t, 3JHF = 9 Hz) and 65% of compound 24 (δ: -80, s).
B: Reaction in the presence of copper in acetonitrile solution.
The reaction is carried out according to the general procedure at the temperature of 25÷30°C. The reaction mixture is analyzed using the method of 19F NMR.
According to the data of 19F NMR, the mixture contains 25% of compound 23 and 75% of compound 24.
The reaction mixture was distilled under reduced pressure (oil pump) collecting volatile products into a trap (-78°C). In the course of distillation, vacuum varied in the range of 0.5-10 torr due to degassing of the distilled mixture. The distillate obtained contains a mixture of products 23, 24, and 25 (the latter is obviously formed as a result of dehydrobromination of compound 24). The mixture is redistilled to give a fraction boiling within the range of 110÷151°C/10 torr. According to 1H and 19F NMR-data the mixture contains 23% of compound 23, 34% of compound 24, and 43% of compound 25, and also a negligible amount of styrene dibromide. Vinylsulfone 25 is crystallized on keeping.
(E)-PhCH=CHSO2CF3 (25).
1H NMR δ: 6.85 (d, 1H, PhCH=), 7.93 (d, 1H, =CHSO2), 7.45-7.65 (m, 5H, Ph); 19F NMR δ: -79 (s, CF3). The mass spectrum (M/Z, reference): 236[M]+, 167[M-CF3]+, 134[C6H5CHCHCF]+, 103[M-SO2CF3]+(100%), 77[C6H5]+, 69[CF3]+, 51[CF2H]+.
References
- F. Ooms, R. Frederick, F. Durant, J.P. Petzer, N. Castagnoli Jr., C. J Van der Schyf, J. Wouters, Bioorg.Med.Chem.Lett., 2003, 13, 69-73.
- J. Reniers, S. Robert, R. Frederick, B. Masereel, S. Vincent, J. Wouters, Bioorg.Med.Chem., 2011, 19, 134-144.
- U. Gerlach, J. Brendel, H.-J. Lang, E.F. Paulus, K. Weidmann, A. Bruggemann, A.E. Busch, H. Suessbrich, M. Bleich, R. Greger, J.Med.Chem., 2001, 44, 3831-3837.
- R.W. Winter, J.X. Kelly, M.J. Smilkstein, R. Dodean, D. Hinrichs, M.K. Riscoe, Exp Parasitol., 2008, 118, 487-497.
- T. Rodrigues, F. Lopes, R. Moreira, Curr.Med.Chem., 2010, 17, 929-956.
- F.A. Romero, S.M. Vodonick, K.R. Criscione, M.J. McLeish, G.L. Grunewald, J.Med.Chem., 2004, 47, 4483-4493.
- M.K. Riscoe, R.W. Winter, J.X. Kelly, M.J. Smilkstein, D.J. Hinrichs, US Pat. № 7,829,578 B1 (2010).
- Q. Yang, B. Lorsbach, G. Whiteker, G. Roth, C. DeAmicis, D.I. Knueppel, A.M. Buysse, K. Gray, X. Li, J.M. Muhuhi, R. Ross Jr., D.E. Podhorez, Y. Zhang, US Pat. № 9,414,594 B2 (2016).
- W. Hierse, N. Ignatyev, M. Seidel, E. Montenegro, P. Kirsch, A. Bathe, US Pat. № 8,067,625 B2 (2011).
- Terasawa, H. Monobe, K. Kiyohara, J.Fluor.Chem., 2006, 127, 954-961.
- Vanover, J.D. Necessary, K.B. Chandalia, J.W. Reisch, J.M. O’Connor, K. Delaney, P.R. Miller, US Pat. № 5,002,678 (1991).
- H. Buchwald, A. Brackmann, B. Raszkowski, US Pat. № 5,304,321 (1994).
- B. Bidlingmeyer, Q. Wang, US Pat. № 7,125,492 B2 (2006).
- K.J. Cross, US Pat. № 8,939,208 B2 (2015).
- M. Antinolo, E. Jimenez, J. Albaladejo, Environ.Sci.Technol., 2011, 45, 4323-4330.
- W.E. Hanford, R.M. Joyce Jr., US Pat. № 2,440,800 (1948).
- M. Van Der Puy, A. Thenappan, US Pat. № 5,777,184 (1998).
- J.-A. Ma, D. Cahard, JFC, 2007, 128, 975-996.
- A. Studer, Angew.Chem.I.E., 2012, 51, 8950-8958.
- S. Barata-Vallejo, Al Postigo, Coord.Chem.Rev., 2013, 257, 3051-3069.
- H. Egami, M. Sodeoka, Chem.Eur.J., 2014, 53, 8294-8308.
- S. Barata-Vallejo, B. Lantano, Al Postigo, Chem.Eur.J., 2014, 20, 16806-16829.
- W. Zeng, F. Chen, Chin.J.Appl.Chem., 2014, 31, 627-641.
- C. Ni, M. Hu, J. Hu, Chem.Rev., 2015, 115, 765-825.
- X.-H. Xu, K. Matsuzaki, N. Shibata, Chem.Rev., 2015, 115, 731-764.
- P. Gao, X.-R. Song, X.-Y. Liu, Y.-M. Liang, Chem.Eur.J., 2015, 21, 7648-7661.
- R.N. Haszeldine, JCS, 1949, 2856-2861.
- N.O. Brace, JOC, 1963, 28, 3093-3102.
- J.D. Park, F.E. Rogers, J.R. Lacher, JOC, 1961, 26, 2089-2095.
- J. Ignatowska, W. Dmowski, JFC, 2007, 128, 997-1006.
- T. Kitazume, N. Ishikawa, JACS, 1985, 107, 5186-5191.
- S. Mukhopadhyay, H.K. Nair, H.S. Tung, M. Van Der Puy, US Pat. № 0245773 A1 (2005).
- V.A. Petrov, C.G. Krespan, JFC, 2000, 102, 199-204.
- G.V.D. Tiers, US Pat. № 2,965,659 (1960).
- N. Kamigata, T. Fukushima, M. Yoshida, J.Chem.Soc.Chem.Commun., 1989, 1559-1560.
- N. Kamigata, M. Yoshida, H. Sawada, M. Nakayama, US Pat. № 5,118,879 (1992).
- H. Jiang, C. Huang, J. Guo, C. Zeng, Y. Zhang, S. Yu, Eur.JOC, 2012, 18, 15158-15166.
- H. Jiang, Y. Cheng, Y. Zhang, S. Yu, Eur.JOC, 2013, 5485-5492.
- S.H. Oh, Y.R. Malpani, N. Ha, Y.-S. Jung, S.B. Han, Org.Lett., 2014, 16, 1310-1313.
- X.-J. Tang, W.R. Dolbier Jr., Angew.Chem.I.E., 2015, 54, 4246-4249.
- D.B. Bagal, G. Kachkovskyi, M. Knorn, T. Rawner, B.M. Bhanage, O. Reiser, Angew.Chem.I.E., 2015, 54, 6999-7002.
- C. Zhang, Adv.Synth.Catal., 2014, 356, 2895-2906.
- Y. Lu, Y. Li, R. Zhang, K. Jin, C. Duan, JFC, 2014, 161, 128-133.
- B. Yang, X.-H. Xu, F.-L. Qing, Org.Lett., 2015, 17, 1906-1909.
- A. Deb, S. Manna, A. Modak, T. Patra, S. Maity, D. Maiti, Angew.Chem.I.E., 2013, 125, 9929-9932.
- D.J. Wilger, N.J. Gesmundo, D.A. Nicewicz, Chem.Sci., 2013, 4, 3160-3165.
- Q. Lefebvre, N. Hoffmann, M. Rueping, Chem.Commun., 2016, 52, 2493-2496.
- W.-Y. Huang, L. Lu, Chin.J.Chem., 1992, 10, 268-273.
- CN Pat. № 1730127 B (2010).
- W.-Y. Huang, J.-L. Chen, ActaChim.Sin.Eng.Ed., 1988, 150-154.
- Y.-F. Zhang, L. Lu, W.-Y. Huang, ActaChim.Sin.Eng.Ed., 1989, 376-384.
- X.-K. Jiang, G.-Z. Ji, J.R.-Y. Xie, JFC, 1996, 79, 133-138.
- L. Chu, F.-L. Qing, Org.Lett., 2012, 14, 2106-2109.
- X. Wu, L. Chu, F.-L. Qing, Angew.Chem.I.E., 2013, 52, 2198-2202.
- F. Wang, X. Qi, Z. Liang, P. Chen, G. Liu, Angew.Chem.I.E., 2014, 53, 1881-1886.
- M. Fu, L. Chen, Y. Jiang, Z.-X. Jiang, Z. Yang, Org.Lett., 2016, 18, 348-351.
- J. Charpentier, N. Fruh, A. Togni, Chem.Rev., 2015, 115, 650-682.
- M. Van Der Puy, T.R. Demmin, G.V.B. Madhavan, A. Thenappan, H.S. Tung, JFC, 1996, 76, 49-54.
- H.K. Nair, A.J. Poss, US Pat. № 6,476,279 B2 (2002).
- Z.-Y. Long, Q.-Y. Chen, Tetrahed.Lett., 1998, 39, 8487-8490.
- M. Huang, L. Li, Z.-G. Zhao, Q.-Y. Chen, Y. Guo, Synthesis, 2015, 47, 3891-3900.
- X.-J. Tang, C.S. Thomoson, W.R. Dolbier Jr., Org.Lett., 2014, 16, 4594-4597.
- G.K.S. Prakash, A.K. Yudin, Chem.Rev., 1997, 97, 757-786.
- R.O. Bolt, R.M. Joyce, Chem.Eng.News, 1947, 25, 1866-1867.
- F. Minisci, Acc.Chem.Res., 1975, 8, 165-171.
- M. Asscher, D. Vofsi, JCS, 1964, 4962-4971.
- L.I. Zakharkin, G.G. Zhigareva, Zh.Org.Khim., 1973, 9, 891-895.
- Y. Amiel, JOC, 1974, 39, 3867-3870.
- N. Roques, US Pat. № 2004/0147789 A1 (2004).
- J.M. Tedder, J.C. Walton, Tetrahedron, 1980, 36, 701-707.
- B. Giese, Angew.Chem.I.E., 1983, 22, 753-764.
Recommended for publication by PhD A.A. Tyutyunov
Fluorine Notes, 2016, 109, 1-2
