Received: September 2017
DOI 10.17677/fn20714807.2017.05.04
Fluorine Notes, 2017, 114, 7-8
Synthesis of 2,2'-azanediylbis(N,N-bis(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl) acetamide) - a novel fluorous secondary amine with four perfluorooctyl chains
Anikó Nemes, Gitta Schlosser, Antal Csámpai, Dénes Szabó and József Rábai*
Institute of Chemistry, Eötvös Loránd University, Pázmány Péter sétány 1-A, 1117 Budapest, Hungary
E-mail: rabai@elte.hu
Abstract: Bis(perfluorooctylpropyl)amine and N-(benzyloxycarbonyl)iminodiacetic acid di(pentafluorophenyl) ester was heated in C6H5CF3 to afford a fluorous diamide with four perfluorooctyl chains, which on deprotection with catalytic hydrogenation in FC-72 (a mixture of perfluorohexanes) solvent gave the appropriate secondary amine in high yield. Although this fluorous secondary amine showed rather low solubility in perfluoroalkanes at room temperature it was found to solubilize colloidal palladium particles in ether, BTF and perfluoroalkanes.
Keywords: fluorous amines, fluorous scavengers, fluorous dendrimers
Fluorous primary (1o), secondary (2o) and tertiary (3o) amines of the type [Rfn(CH2)mRfn]xNH3-x) are important building blocks and reagents in fluorous chemistry [1].Their fluorous aldehyde and fluorous alkyl iodide precursors are easily accessible [2].The 2o amine [(C6F13CH2CH2)3SiCH2CH2CH2]2NH with six perfluorohexyl chains was synthesized and introduced as a fluorous scavenger in the automated solution phase parallel synthesis of an urea library by Curran et al. [3]. Another application of a heavy fluorous support with six perfluorooctyl chains (Scheme 1), enabled the synthesis of a bio-active peptide, Leu-enkephalin using Fmoc-strategy as reported by Mizuno et al. [4].

Figure 1. Structure of a heavy fluorous support used for peptide synthesis (cf. [4]).
The synthesis of a novel 2o fluorous amine having four perfluorooctyl groups (6) is disclosed here, which involves the reaction of bis(perfluorooctylpropyl)amine (4) and N-(benzyloxycarbonyl)iminodiacetic acid di(pentafluorophenyl)ester (3) followed by deprotection of the intermediate fluorous carbamate (5) using catalytic hydrogenation in FC-72 solvent [5] (Scheme 2).
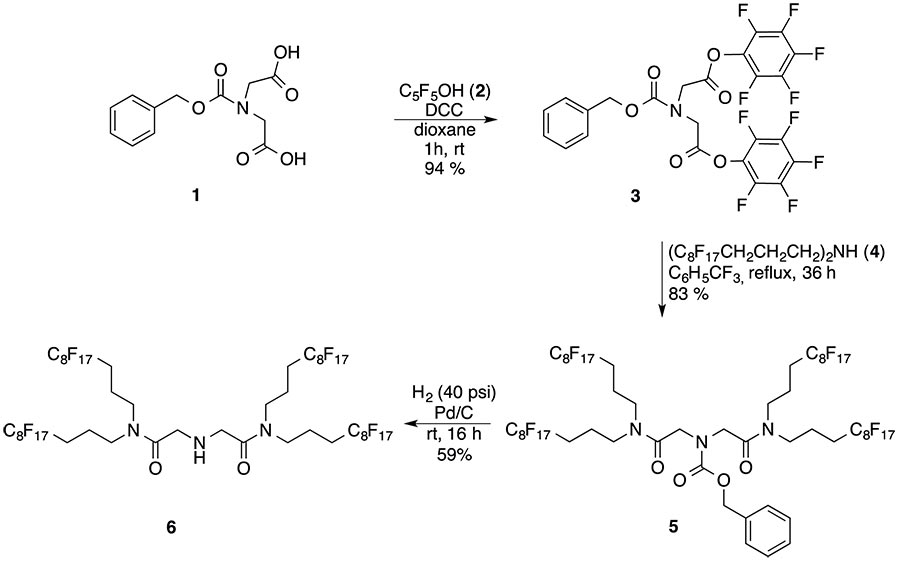
Scheme 2. Synthesis of a dendrimer type fluorous 2o amine 6 with four perfluorooctyl-chains.
However, the synthesis of the precursor active ester 3 was optimized to afford it in 94% yield by using recrystallized N-protected iminodiacetic acid 1 along with freshly distilled pentafluorophenol 2 and excluding air humidity. The reaction of 3 (1 mol) and amine 4 (2 mol) took place under homogeneous condition in benzotrifluoride (BTF) to afford 5 -the protected diamide with four perfluorooctyl groups in 83% yield - as a pale yellow oil after a fluorous-organic biphasic extractive work-up. Carbamate 5 (%F = 61.35) showed high solubility in perfluoroalkanes, while the solid fluorous amine 6 (%F= 65.52), obtained by the Pd/C catalyzed hydrogenolysis of 5, disclosed relatively low fluorous solubility and a melting point of 99-101oC. This behavior is in accordance with the empirical principles which predict that intermolecular attractive interactions should be minimized to achieve higher fluorophilicity values and fluorocarbon solubility [6]. Consequently amine 6 in spite of its higher fluorine percent than the precursor carbamate 5, does not meet the requirements for application as a fluorous scavenger [7]. However, compound 6 may be used as an F-building block or as a reagent to stabilize and solubilize Pd-colloids in perfluoroalkanes. A small amount of the mixture of the precipitated 6 and Pd/C was heated with a few mL of perfluoro(methylcyclohexane) (CF3C6F11) for a minute and filtered to give a brilliant but brown colored solution. This observation is the indication of the formation a fluorocarbon soluble palladium-sol. When this solution was shaken with Ph3P in toluene a colorless CF3C6F11 layer was obtained, but soon a black precipitate appeared at the phase boundary. In a different experiment the Pd-sol decomposed completely by boiling it for a few minutes.
Experimental
The precursors 1 and 4 were prepared as reported in [8] and [1b], respectively. Fluorinert FC-72 electronic fluid, BTF and CF3CH2OH was purchased from FC Chemicals, while the other reagents and organic solvents from Sigma-Aldrich and MOLAR. 1H-, 13C- and 19F-NMR spectra were recorded on Bruker Avance 250 instrument using a 5 mm inverse 1H/13C/31P/19F probe head at room temperature. Chemical shifts (δ) are given in parts per million (ppm) units relatively solvent (CDCl3) residual peaks (δ=7.26 for 1H, δ=77.0 for 13C) and to CFCl3 as external standard (δ=0.00 for 19F). Determination of molecular mass and acquisition of the tandem mass spectrum were performed by electrospray ionization mass spectrometry (ESI-MS) on a Bruker Daltonics Esquire 3000 plus (Germany) ion trap mass spectrometer. The sample was dissolved in acetonitrile-trifluoroethanol solvent mixture (50:50, V/V). The mass spectrum was acquired in the 200-3000 m/z range. Capillary voltage was 4000 V, nebulizer gas pressure was 10 psi, drying gas flow was 4 L/min and the heated capillary temperature was 250 oC. The sample was injected into the ion source in a flow rate of 10 L/min using a syringe pump. Melting points were determined on a Böetius micro-melting point apparatus and are uncorrected. Gas chromatographic analysis of volatile products was performed using a Hewlett-Packard 5890 Series II instrument with PONA [crosslinked methylsilicone gum] 50 m x 0.2mm x 0.5 mm column, H2 carrier gas, FID detection; Program: 120 °C, 5 min, 10 °C/min, 250 °C, 5 min, Inj.: 250°C, Det.: 280°C.
Bis(pentafluorophenyl) 2,2'-(((benzyloxy)carbonyl)azanediyl)diacetate (3)
In
a flame dried flask under and argon atmosphere to a magnetically stirred mixture of N-(benzyloxycarbonyl)-iminodiacetic
acid(1) (3.00g, 11.2 mmol) and C6F5OH (2)
(4.58 g, 24.6 mmol) in absolute dioxane (60 mL) was added dropwise a solution of DCC (5.07 g, 24.6
mmol) in absolute dioxane (12 mL) at room temperature during 1 h.The progress of reaction was indicated
with the formation of a thick white precipitate (DCU). The mixture was kept at room temperature for
overnight, than the precipiate was removed by filtration. It was washed with dioxane (3 × 10 mL)
and dried to give 5.05 g (22.6 mmol, 92%) of DCU side product. The filtrate was evaporated using
a rotavapor at 16 mmHg and 50 oC bath temperature for 30 min to afford 8.2 g of crude
melt, which was solidified on cooling to room temparature. Recrystallization from n-heptane
(50 mL) gave 5.10 g (76%) of white crystalline product with m.p.= 80-82oC. Upscaling this
synthesis by 4 times (i.e. using 12.0 g of 1 [C6H5CH2OC(O)N(CH2CO2H)2])
gave 25.2 g (94%) of product of the same purity as earlier, which showed agreeable spectral properties
to that reported in [9].
1H-NMR (CDCl3) (, ppm): 7.29, s, (5H, C6H5); 5.17, s, (2H, PhCH2); 4.46, s, (NCH2C=O, A-chain); 4.55, s, (NCH2C=O, B-chain).
13C-NMR (CDCl3) (, ppm): 128.9 (Car-4); 129.0 (Car-3,5); 128.5 (Car-2,6); 135.7 (Car-1); 69.3 (CH2OCO); 155.8 (CH2OCO); 49.1 (CH2CO2, A-chain); 165.9 (CH2CO2, A-chain); 49.4 (CH2CO2, B-chain); 166.0 (CH2CO2, B-chain).
Benzyl bis(2-(bis(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl)amino)-2-oxoethyl) carbamate (5)
Under an argon atmosphere a stirred mixture of 4 (7.12 g, 7.60 mmol) and active ester 3 (2.28 g, 3.80 mmol) in C6H5CF3 (30 mL) was refluxed on a 130oC oil-bath until the GC analysis of a sample showed no more increase in the 2:4 ratio. This approximately took 36 h. The solvent was evaporated in vacuum (rotavapor) then the residual oil was dissolved in an FC-72 (35 mL) – CH3OH biphasic system. The side product C6F5OH showed a preference to the CH3OH phase. The lower fluorous phase was separated and consecutively washed with CH3OH (2 × 15 mL) and 2M NaOH (2 × 50 mL). The phase separation, which indicated with the clearing up of the upper aqueous phase, was facilitated with addition of some drops of CH3OH. The FC-72 phase was washed again with CH3OH (2 × 15 mL), then it was separated and dried (Na2SO4). The filtrate was concentrated by distillation off the solvent at atmospheric pressure to allow the recovery most of FC-72 (25 mL) solvent. The last traces of FC-72 from the residual oil were removed at 16 Hgmm pressure and 110oC temperature for 1 h. Yield: 6.65 g (83%) pale yellow oil. Compound 5 is completely miscible with perfluoroalkanes, benzotrifluoride, and ether. It is free from 2 and 4 as determined by GC.
1H-NMR (CF2ClCFCl2-CDCl3, 1:1 v/v, TMS) (, ppm): 4.17, s, (2H, NCH2CO-, A-chain); 4.25 s, (2H, NCH2CO-, B-chain); 5.04 s, (2H, PhCH2OCO-); 7.13,~s, (5H, C6H5CH2).
13C-NMR(CF2ClCFCl2-CDCl3, 1:1 v/v, TMS) (, ppm): 136.3 (Car-1); 128.5 (Car-2, 6); 128.7 (Car-3,5); 128.6 (Car-4); 68.5 (PhCH2); 156.4 (PhCH2OC=O); 48.7 (CH2C(=O)N, A-chain); 168.6 (CH2C(=O)N, A-chain); 47.1 (NCH2CH2, A-1-chain); 45.7 (NCH2CH2, A-2-chain); 49.4 (CH2C(=O)N, B-chain); 169.1 (CH2C(=O)N, B-chain); 46.7 (NCH2CH2, B-1-chain); 45.8 (NCH2CH2, B-2-chain).
2,2'-Azanediylbis(N,N-bis(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl)acetamide) (6)
The 250 mL volume glass vessel of a medium pressure autoclave was charged with carbamate 5 (6.74 g, 3.2 mmol) dissolved in FC-72 (60 mL) and 10% Pd/C catalyst (600 mg Pd-C). Then the mixture was hydrogenated at 40 psi pressure of H2 at room temperature for 16 h. The crude product was isolated by filtration, washed with FC-72 and dried under vacuum. Yield: 6.27 g (~ 90%) off-white solid (6@Pd/C). This crude product was extracted with boiling ether in a Soxhlet apparatus for 48 h. The extract was evaporated on a rotavapor then the crystalline product obtained was filtered, washed with CH3OH and dried over P2O5 in a vacuum. Yield: 3.69 g (59%) white crystalline product, m.p. = 99-101oC.
1H-NMR (CF2ClCFCl2-CDCl3, 1:1 v/v, TMS) (, ppm): 3.51, s, (2H, NCH2CO); 3.45, t, (2H, 7.6 Hz, NCH2CH2, A-chain); 3.37, t, (2H, 7.6 Hz, NCH2CH2, B-chain); 2.12, br, (4H, NCH2CH2, A-chain and B-chain); 1.92, tt, (4H, 14.4 Hz, 7.3 Hz, A-chain and B-chain).
13C-NMR: 50.5 (H-NCH2); 171.2 (C=O); 45.6 (N-CH2CH2CH2, A-chain); 20.6 (N-CH2CH2CH2, A-chain); 29.1, t, (N-CH2CH2CH2C8F17, A-chain); 46.9 (N-CH2CH2CH2, B-chain); 19.5 (N-CH2CH2CH2, B-chain); 28.7, t, (N-CH2CH2CH2C8F17, B-chain).
MS(ESI) (CH3CN : CF3CH2OH = 1:1 v/v): Calcd. forC48H29F68N3O2 =
1971.1; Observed: 1971.1
(Cf. Supporting Information).
Acknowledgement
We thank the National Research, Development and Innovation Office for the financial support of the M-ERA.Net COR_ID program (NKFIH NN117633). G. S. acknowledges the support by the MTA János Bolyai Research Scholarship and by the MTA Premium Post-Doctorate Research Program of the Hungarian Academy of Sciences (HAS, MTA).
Supporting Information
The MS spectrum of compound 6 is disclosed.
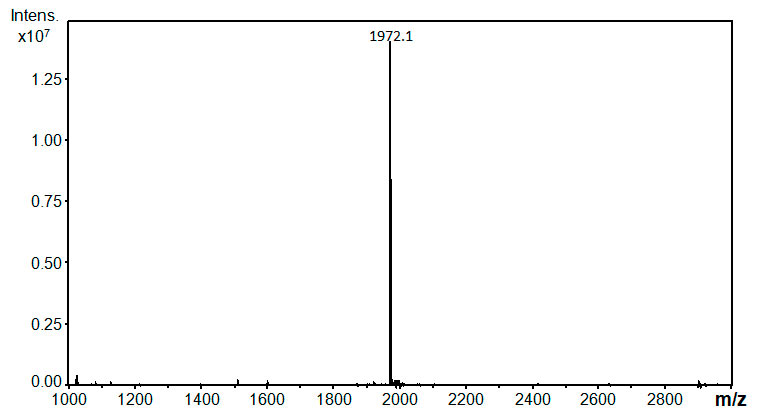
Figure S1. ESI-MS spectrum of 6 acquired on a Bruker Daltonics Esquire 3000 plus (Germany) ion trap mass spectrometer. The sample was dissolved in acetonitrile-trifluoroethanol solvent mixture (50:50, V/V). The detected ion corresponds to the protonated molecule [M+H]+.
References
- (a) C. Rocaboy, W. Bauer, and J.A. Gladysz, Convenient Syntheses of a Family
of Easily Recoverable Fluorous Primary, Secondary, and Tertiary Aliphatic Amines NH3-x[(CH2)m(CF2)7CF3]x(m=
3-5; x=1, 2, 3) - FineTuning of Basicities and Fluorous Phase Affinities, Eur. J. Org. Chem. 2000,
2621-2628;
DOI: 10.1002/1099-0690(200007)2000:14<2621::AID-EJOC2621>3.0.CO;2-H
(b) Z. Szlávik, G. Tárkányi, Á. Gömöry, Gy. Tarczay, J. Rábai,Convenient syntheses and characterization of fluorophilic perfluorooctyl-propylamines and ab initio calculations of proton affinities of related model compounds, J. Fluorine Chem.2001, 108, 7-14.
https://doi.org/10.1016/S0022-1139(00)00398-5
(c) Szabó, D.; Mohl, J.; Bálint, A.-M.; Bodor, A.; Rábai, J. Novel generation ponytails in fluorous chemistry: Synthesis of primary, secondary, and tertiary (nonafluoro-tert-butyloxy)ethyl amines. J. Fluorine Chem.2006,127, 1496-1504; DOI: 10.1016/j.jfluchem.2006.06.020 - (a) Nemes, A.; Berta, M.; Ivanko, P.; Szabó, D.; Rábai, J. Synthesis of 3-perfluoroalkylpropanals
and 3-perfluoroalkylpropionitriles. FLUORINE NOTES / FTORNIE ZAMETKI: 6(103):November-December (2015);
DOI 10.17677/fn20714807.2015.06.02;
(b) Menczinger, B.; Jakab, G.; Szabó, D.; Rábai, J. Synthesis of 1-iodo-3-perfluoroalkylpropanes and 1-iodo-4-perfluoroalkylbutanes. FLUORINE NOTES / FTORNIE ZAMETKI: (3) 94. May-June (2014). - Linclau, B., Singh, A.K. and Curran, D.P., Organic-fluorous phase switches: A fluorous amine scavenger for purification in solution phase parallel synthesis.J. Org. Chem.,1999, 64, 2835–2842. DOI:10.1021/jo9823442
- Mizuno, M.; Goto, K.;Miura, T.; Matsuura, T.; Inazu, T. Peptide synthesis on fluorous support.Tetrahedron Lett.2004, 45, 3425-3428. DOI: 10.1016/j.tetlet.2004.03.013
- http://multimedia.3m.com/mws/media/64892O/fluorinert-electronic-liquid-fc-72.pdf (accessed 03.08.2017)
- Kiss, L. E.; Kövesdi, I., Rábai, J. An Improved Design of FluorophilicMolecules: Prediction of the ln P Fluorous Partition Coefficient, Fluorophilicity, Using 3D QSAR Descriptors and Neural Networks. J. Fluorine Chem.2001,108,95-109.https://doi.org/10.1016/S0022-1139(01)00342-6
- (a) Lindsley, C. W.;Zhao, Z.; Leister, W. H.; Strauss, K. A. Fluorous-tethered amine bases for organic
and parallel synthesis: scope and limitations. Tetrahedron Letters,2002,
43, 6319-6323. https://doi.org/10.1016/S0040-4039(02)01399-0
(b) Chen, H.-T. C.; Zhang, W. Fluorous reagents and scavengers versus solid-supported reagents and scavengers, a reaction rate and kinetic comparision. Molecular Diversity, 2005, 9, 353-359. DOI: 10.1007/s11030-005-8104-3 - Abalain, C.; Langlois, M., Synthesis of dibenzyliminodiaceticacid derivatives as potential inhibitors of HIV-1 aspartyl protease. Eur. J. Med. Chem. Chim. Ther., 33 (2) 1998, 155-160. https://doi.org/10.1016/S0223-5234(98)80041-X
- (a) Lutter,
H.-D., Diederich, F. Synthesis of a macrobicyclic thiazolium-host and supramolecular catalysis of
the benzoin condensation. Angew. Chem., GE, 98 (12) 1986, 1125-1127; DOI: 10.1002/anie.198611251
(b) Diederich, F.; Schurmann, G.; Chao, I., Designed water-soluble macrocyclic esterases: from nonproductive to productive binding. J. Org. Chem.,53 (12), 1988, 2744-2757. DOI: 10.1021/jo00247a017
Recommended for publication by Prof. József Rábai
Fluorine Notes, 2017, 114, 7-8
