Received: October 2017
DOI 10.17677/fn20714807.2017.06.02
Fluorine Notes, 2017, 115, 3-4
Photocatalytic radical addition of 1,1,2,2-tetrafluoro-1-iodoethane to alkenes
Svatava Voltrová and Petr Beier*
Institute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo nám. 542/2, 166 10 Prague 6, Czech Republic
E-mail: beier@uochb.cas.cz
Abstract: Photocatalytic radical addition and addition-elimination reactions of terminal alkenes with 1,1,2,2-tetrafluoro-1-iodoethane under a blue LED light irradiation are reported. The photoredox catalysis with Rhodamine B or Eosin Y provided the corresponding 1,1,2,2-tetrafluoro-4-iodoalkanes or 1,1,2,2-tetrafluoroalkenes, respectively, in good yields. The obtained iododerivatives were successfully hydrodeiodinated to form saturated 1,1,2,2-tetrafluoroalkanes.
Keywords: photocatalysis; radical addition; Rhodamine B; Eosin Y; tetrafluoroiodoethane; LED
Introduction
Radical perfluoroalkylation is a popular method for the incorporation of perfluoroalkyl groups into organic molecules with a purpose of influencing properties of compounds [1-2]. Traditionally, perfluoroalkyl halides were used for the generation of perfluoroalkyl radicals; however, more recently metal sulfonates and sulfonyl chlorides have proven to be suitable perfluoroalkyl radical precursors [3-7]. Various approaches for homolytic cleavage of the carbon-halogen bond of perfluoroalkyl halides have been reported, these include radical initiators (AIBN [8], Et3B [9], or peroxide [10]), metals (magnesium [11], iron [1], or copper [12]), Na2S2O4 [13], catalytic triphenylphosphine [14], or UV light [15-16]. Recently, photoredox catalysis and in particular visible-light-promoted perfluoroalkylation [7, 17-20] have emerged as alternatives to initiator-promoted radical perfluoroalkylations. In this field, organic dyes enjoy an advantage over expensive ruthenium or iridium photocatalysts. Here we report Rhodamine B or Eosin Y catalyzed photocatalytic radical addition or addition-elimination, respectively, of HCF2CF2I to terminal alkenes. Compounds with the tetrafluoroethyl group are rare [21-26] and this work should improve access to them in order to study their properties.
Results and discussion
In this work, we have used a blue LED light excited Rhodamine B [27] or Eosine Y [28] as photocatalysts in radical addition or addition-elimination reactions of HCF2CF2I with alkenes. Terminal alkenes 1 bearing different functional groups (alkyl, aryl, halogen, carbonyl, epoxy, hydroxyl, carboxylic acid) underwent radical addition of only equimolar amount of 1,1,2,2-tetrafluoro-1-iodoethane (2) in the presence of (i-Pr)2NEt and catalytic Rhodamine B in water to form the corresponding tetrafluoroiodoalkanes 3 in high yields (Scheme 1). Reactions proceeded smoothly at ambient temperature and complete conversion of 1 was reached under 16 hours (GC-MS control). Interestingly, an experiment with 3b revealed that a green LED light (525 nm) could be used with similar efficiency to the blue light. Under the conditions shown in Scheme 1, hex-1-yne and cis-stilbene did not lead to the addition products.
Scheme 1
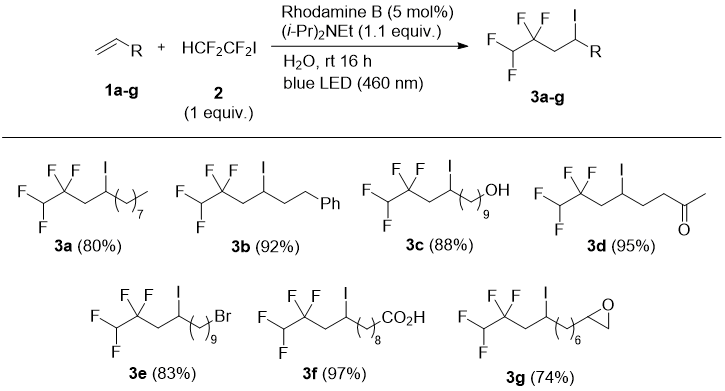
The 19F NMR spectra of iodo derivatives 3 showed an interesting splitting patterns owing to the presence of magnetically non-equivalent (diastereotopic) fluorine atoms (Figure 1).
Figure 1 19F {1H} NMR spectrum of 3a
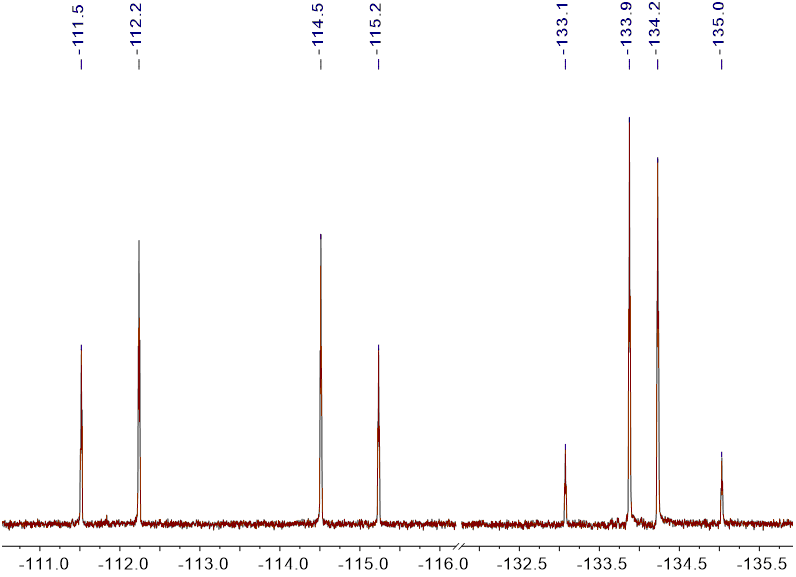
It can be expected that the mechanism operating involves a homolytic cleavage of the carbon-iodine bond and the formation of radicals which add to terminal alkenes. In order to probe this mechanism a reaction with diethyl 2,2-diallylmalonate (4) was performed. The formed fluoroalkyl radical induced a cascade radical addition – 5-exo-trig cyclization to the cyclopentane product 5, which was isolated in good yield and high cis-selectivity (Scheme 2).
Scheme 2
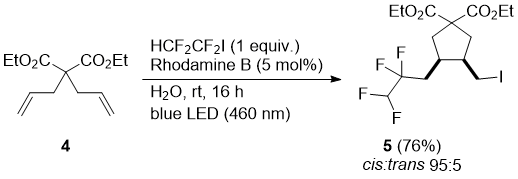
Diethyl allylmalonate (6) afforded under the Rhodamine B catalysis mostly alkene 7a (product of HI elimination after radical addition) in a mixture with addition product 7b. However, using Eosin Y as a photocatalyst, clean formation of 7a was observed (Scheme 3). The formation of the elimination product is in an agreement with previous report using Eosin Y and cesium carbonate [28].
Scheme 3
Next, we turned our attention to substitution of iodine atom of compounds 3 for hydrogen in order to access terminal tetrafluoroethyl-substituted aliphatic compounds. Towards this goal, compound 3b was treated with excess LiAlH4 in THF to form (5,5,6,6-tetrafluorohexyl)benzene (8) in high yield (Scheme 4). This reaction was found to be substrate dependent: when epoxide 3g was reduced either with LiAlH4 or with H2/Pd, no reaction was observed even after prolonged reaction time (48 h) and the starting compound was isolated unchanged. The 19F NMR spectrum of 8 contains two set of signals: a doublet (2JFH = 54.0 Hz) for the CF2H group and a triplet of doublets (3JFH = 18.2, 2.0 Hz) for the CF2 group (Figure 2).
Scheme 4
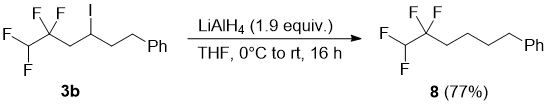
Figure 2. 19F NMR spectrum of 8; 19F {1H} spectrum is shown as an insert
Conclusion
In conclusion, a combination of inexpensive organic dye catalyst and LED light radical addition of tetrafluoroiodoethane to terminal alkenes afforded 1,1,2,2-tetrafluoro-4-iodoalkanes in high yields. Alternatively, radical addition and subsequent HI elimination provided the corresponding 1,1,2,2-tetrafluoroalkenes. The iodo derivative was hydrodeiodinated with lithium aluminum hydride to form saturated 1,1,2,2-tetrafluoroalkane. Thus, tetrafluoroiodomethane (HCF2CF2I) proved to be a useful and general reagent for radical introduction of the HCF2CF2 group under photocatalytic conditions.
Experimental part
General. Reactions with air-sensitive materials were carried out under argon atmosphere using standard Schlenk techniques. All commercially available chemicals were used as received unless stated otherwise. Flash column chromatography was performed using silica gel 60 (0.040–0.063 mm). 1H, 13C, and 19F NMR spectra were measured at ambient temperature using 5 mm diameter NMR tubes. 13C NMR spectra were proton decoupled. The chemical shift values (δ) are reported in ppm relative to internal Me4Si (0 ppm for 1H and 13C NMR) or residual solvents and internal CFCl3 (0 ppm for 19F NMR). Coupling constants (J) are reported in Hertz. GC-MS spectra were recorded using a 5% phenyl methylpolysiloxane column coupled with a quadrupole mass-selective electron impact (EI) detector (70 eV). High resolution MS spectra (HRMS) were recorded using electrospray (ESI), electron impact (EI) or dual ESI/MALDI ionizations. A 3 W LED sources (blue 455-475 nm or green 515-535 nm) containing a light-conductive glass rod immersed directly into the reaction vessel were used.
Synthesis of compounds 3. A mixture of alkene 1 (1.0 mmol) and water (10 mL) was cooled to 0 °C and degassed using three pump-thaw cycles under nitrogen atmosphere. To this emulsion were added (i-Pr)2NEt (192 μL, 1.10 mmol), Rhodamine B (24 mg, 0.05 mmol), and HCF2CF2I (227 mg, 103 μL, 1.0 mmol) using a pre-cooled syringe at 0 °C. The stirred reaction mixture was warmed to rt and irradiated with a blue LED lamp for 16 h (GC-MS control). The reaction mixture was then diluted with Et2O, washed with water and brine. The organic layer was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:EtOAc, 97:3) to afford product 3.
1,1,2,2-Tetrafluoro-4-iodododecane (3a): Yield: 80%; colorless oil, 1H NMR (400 MHz, CDCl3) δ 5.72 (tdd, J = 53.9, 3.0, 2.3 Hz, 1H), 4.39–4.27 (m, 1H), 2.96–2.57 (m, 2H), 1.91–1.69 (m, 2H), 1.61–1.38 (m, 2H), 1.36–1.21 (m, 10H), 0.89 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, CDCl3) δ 117.52 (tt, J = 249.2, 29.4 Hz), 110.03 (tt, J = 249.9, 41.1 Hz), 40.85 (t, J = 21.2 Hz), 40.64, 31.99, 29.73, 29.51, 29.37, 28.71, 22.81, 22.41 (t, J = 2.0 Hz), 14.24; 19F NMR (377 MHz, CDCl3) δ –112.75 to –117.32 (m, 2F), –134.35 to –137.52 (m, 2F).; HRMS (EI+) m/z calcd for C12H21F4I [M]+: 368.0624, found 368.0621.
(5,5,6,6-Tetrafluoro-3-iodohexyl)benzene (3b): Yield: 92%; colorless oil, 1H NMR (400 MHz, CDCl3) δ 7.35–7.26 (m, 2H), 7.27–7.17 (m, 3H), 5.70 (tdd, J = 53.9, 3.0, 2.2 Hz, 1H), 4.28 (tt, J = 8.4, 5.3 Hz, 1H), 3.00–2.63 (m, 4H), 2.13 (tt, J = 7.9, 5.0 Hz, 2H); 13C NMR (101 MHz, CDCl3) δ 140.25, 128.71, 128.66, 126.46, 117.50 (tt, J = 249.5, 29.6 Hz), 109.97 (tt, J = 250.1, 41.1 Hz), 42.06 (d, J = 2.1 Hz), 40.85 (t, J = 21.2 Hz), 35.84, 21.52 (t, J = 2.5 Hz).; 19F NMR (376 MHz, CDCl3) δ –111.96 to –116.66 (m, 2F), –133.82 to –136.55 (m, 2F); HRMS (EI+) m/z calcd for C12H13F4I [M]+: 359.9998, found 359.9994.
12,12,13,13-Tetrafluoro-10-iodotridecan-1-ol (3c): Yield: 88%; colorless oil, 1H NMR (400 MHz, CDCl3) δ 5.71 (tt, J = 53.9, 2.7 Hz, 1H), 4.31 (tt, J = 8.6, 5.1 Hz, 1H), 3.61 (t, J = 6.2 Hz, 2H), 2.89–2.58 (m, 2H), 2.02 (s, 1H), 1.84–1.70 (m, 2H), 1.59–1.49 (m, 2H), 1.40–1.21 (m, 12H); 13C NMR (101 MHz, CDCl3) δ 117.46 (tt, J = 249.1, 29.4 Hz), 109.96 (tt, J = 250.0, 41.0 Hz), 63.07, 40.78 (t, J = 21.1 Hz), 40.53 (d, J = 1.9 Hz), 32.86, 29.64, 29.57, 29.46, 29.40, 28.60, 25.82, 22.39; 19F NMR (376 MHz, CDCl3) δ –113.20 to –117.20 (m), –134.18 to –137.30 (m); HRMS (ESI+) m/z calcd for C13H24F4IO [M+H]+: 399.0808, found 399.0811.
7,7,8,8-Tetrafluoro-5-iodooctan-2-one (3d): Yield: 95%; colorless oil, 1H NMR (400 MHz, CDCl3) δ 5.74 (tdd, J = 53.8, 3.2, 2.2 Hz, 1H), 4.49–4.23 (m, 1H), 2.99–2.55 (m, 4H), 2.17 (s, 3H), 2.16–2.07 (m, 1H), 1.97 (dddd, J = 15.2, 9.8, 8.1, 5.4 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 206.79, 117.34 (tt, J = 249.4, 29.4 Hz), 109.92 (tt, J = 250.1, 40.8 Hz), 43.75, 41.03 (t, J = 21.3 Hz), 34.25 (d, J = 2.2 Hz), 30.21, 21.21 (t, J = 2.7 Hz); 19F NMR (376 MHz, CDCl3) δ –112.26 to –116.45 (m), –134.10 to –136.77 (m); HRMS (ESI+) m/z calcd for C8H12F4IO [M+H]+: 326.9869, found 326.9866.
13-Bromo-1,1,2,2-tetrafluoro-4-iodotridecane (3e): Yield: 83%; colorless oil, 1H NMR (400 MHz, CDCl3) δ 5.72 (tdd, J = 53.9, 3.1, 2.2 Hz, 1H), 4.32 (tdd, J = 8.3, 5.8, 4.3 Hz, 1H), 3.40 (t, J = 6.8 Hz, 2H), 2.92–2.55 (m, 2H), 1.90–1.80 (m, 4H), 1.36–1.23 (m, 12H); 13C NMR (101 MHz, CDCl3) δ 117.48 (tt, J = 249.1, 29.4 Hz), 114.26, 109.98 (tt, J = 250.0, 41.0 Hz), 40.81 (t, J = 21.2 Hz), 40.54 (d, J = 1.9 Hz), 34.11, 32.93, 29.65, 29.42, 29.37, 28.81, 28.59, 28.26, 22.37; 19F NMR (376 MHz, CDCl3) δ –112.92 to –117.43 (m), –134.50 to –137.09 (m); HRMS (EI+) m/z calcd for C13H21F4BrI [M–H]+: 458.9808, found 368.9799.
12,12,13,13-Tetrafluoro-10-iodotridecanoic acid (3f): Yield: 97%; colorless oil, 1H NMR (400 MHz, CDCl3) δ 5.72 (tt, J = 53.9, 2.7 Hz, 1H), 4.32 (tdd, J = 8.3, 5.7, 4.3 Hz, 1H), 2.95–2.55 (m, 2H), 2.35 (t, J = 7.5 Hz, 2H), 1.79 (ddp, J = 19.7, 9.8, 4.5 Hz, 2H), 1.63 (p, J = 7.4 Hz, 2H), 1.53 (tq, J = 7.7, 3.8, 2.8 Hz, 1H), 1.42–1.29 (m, 9H); 13C NMR (101 MHz, CDCl3) δ 180.37, 117.51 (tt, J = 249.1, 29.4 Hz), 110.01 (tt, J = 250.1, 41.1 Hz), 40.81 (t, J = 21.1 Hz), 40.54 (d, J = 1.9 Hz), 34.18, 29.65, 29.27, 29.24, 29.10, 28.57, 24.75, 22.37 (t, J = 2.5 Hz); 19F NMR (376 MHz, CDCl3) δ –112.70 to –117.55 (m), –134.26 to –137.53 (m); HRMS (ESI+) m/z calcd for C13H22F4IO2 [M+H]+: 413.0601, found 413.0602.
2-(9,9,10,10-Tetrafluoro-7-iododecyl)oxirane (3g): Yield: 74%; colorless oil, 1H NMR (400 MHz, CDCl3) δ 5.71 (tdd, J = 53.9, 3.2, 2.2 Hz, 1H), 4.31 (tdd, J = 8.3, 5.7, 4.3 Hz, 1H), 2.89 (tdd, J = 5.0, 3.9, 2.7 Hz, 1H), 2.86–2.60 (m, 2H), 2.77–2.68 (m, 1H), 2.45 (dd, J = 5.0, 2.7 Hz, 1H), 1.78 (ddt, J = 14.7, 9.6, 5.0 Hz, 2H), 1.58–1.22 (m, 10H); 13C NMR (101 MHz, CDCl3) δ 117.48 (tt, J = 249.2, 29.4 Hz), 109.97 (tt, J = 250.1, 41.0 Hz), 52.41, 47.18, 40.79 (t, J = 21.2 Hz), 40.46 (d, J = 1.8 Hz), 32.52, 29.57, 29.28, 28.54 (d, J = 1.8 Hz), 25.99, 22.27 (t, J = 2.5 Hz); 19F NMR (376 MHz, CDCl3) δ –112.73 to –117.45 (m), –134.39 to –137.12 (m); HRMS (ESI+) m/z calcd for C12H20F4IO [M+H]+: 383.0495, found 383.0493.
Synthesis of compound 5, (diethyl cis-3-(iodomethyl)-4-(2,2,3,3-tetrafluoropropyl)cyclopentane-1,1-dicarboxylate): Diethyl 2,2-diallylmalonate (4) (120 mg, 0.5 mmol) and water (5 mL) were cooled to 0 °C and degassed using three pump−thaw cycles under nitrogen atmosphere. To this emulsion was added Rhodamine B (12 mg, 0.025 mmol) and then iodide 2 (227 mg, 103 μL, 1.0 mmol) by pre-cooled syringe at 0 °C. The stirred reaction mixture was warmed to rt and irradiated with a blue LED lamp for 16 h (GC-MS control). The reaction mixture was then diluted with Et2O and washed with water and brine. The organic phase was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:EtOAc, 99:1) to afford product 5 (180 mg of yellow oil, 76% yield) as the mixture of cis:trans isomers in ratio 95:5. 1H NMR (400 MHz, CDCl3) δ 5.70 (tt, J = 54.0, 2.6 Hz, 1H), 5.11–5.07 (m, 1H), 5.06 (t, J = 1.2 Hz, 1H), 4.26–4.08 (m, 4H), 3.18 (ddd, J = 9.8, 5.2, 1.0 Hz, 1H), 3.01 (t, J = 9.9 Hz, 1H), 2.61 (dt, J = 7.5, 1.2 Hz, 2H), 2.57–2.43 (m, 2H), 2.34–2.18 (m, 1H), 2.14–1.82 (m, 1H), 1.28–1.15 (m, 6H); 13C NMR (101 MHz, CDCl3) δ 172.03, 170.70, 132.30, 119.09, 117.85 (tt, J = 247.3, 29.5 Hz), 110.11 (tt, J = 249.4, 41.6 Hz), 61.76 (d, J = 9.6 Hz), 61.19, 45.44, 39.73, 36.70, 35.41 (d, J = 2.2 Hz), 28.45 (t, J = 21.8 Hz), 14.09, 13.97 (d, J = 3.0 Hz); 19F NMR (376 MHz, CDCl3) δ –102.18 to –108.83 (m), –125.64 (ddd, J = 53.3, 38.9, 8.5 Hz); HRMS (ESI+) m/z calcd for C15H22F4IO4 [M+H]+: 469.04934, found 469.04935.
Addition of HCF2CF2I to diethyl 2-allylmalonate6:
a) Reaction catalyzed with Rhodamine B. A mixture of diethyl 2-allylmalonate (6) (200 mg, 1 mmol) and water (10 mL) was cooled to 0 °C and degassed using three pump−thaw cycles under nitrogen atmosphere. To this emulsion was added (i-Pr)2NEt (192 μL, 1.10 mmol), Rhodamine B (24 mg, 0.050 mmol), and HCF2CF2I (227 mg, 103 μL, 1.0 mmol) by pre-cooled syringe at 0 °C. The stirred reaction mixture was warmed to rt and irradiated with a blue LED lamp for 16 h. The reaction mixture contained according to the GC-MS analysis a mixture of the addition-elimination product 7a and addition product 7b. The dark-red suspension was diluted with Et2O, washed with water and brine. The organic phase was dried (MgSO4) and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography (hexane:EtOAc, 95:5) to afford a mixture 7a and 7b (250 mg of dark yellow oil, 58% yield, 7a:7b 4:1).
b) Reaction catalyzed with Eosin Y. A mixture of diethyl 2-allylmalonate (6) (200 mg, 1 mmol) and water (10 mL) was cooled to 0 °C and degassed using three pump−thaw cycles under nitrogen atmosphere. To this emulsion was added Cs2CO3 (652 mg, 2 mmol), Eosin Y (32 mg, 0.05 mmol), and HCF2CF2I (454 mg, 206 μL, 2.0 mmol) by pre-cooled syringe at 0 °C. The stirred reaction mixture was warmed to rt and irradiated with a blue LED lamp for 20 h. The reaction mixture contained according to the GC-MS analysis pure addition-elimination product 7a. The red suspension was diluted with Et2O and washed with water and brine. The organic phase was dried (MgSO4) and concentrated under reduced pressure to yield 247 mg (82%) of colorless oil 7a.
Diethyl (E)-2-(4,4,5,5-tetrafluoropent-2-en-1-yl)malonate (7a): 1H NMR (400 MHz, CDCl3) δ 5.75 (tt, J = 53.8, 2.9 Hz, 1H), 4.34–4.07 (m, 4H), 2.40–2.21 (m, 1H), 2.15–2.00 (m, 1H), 1.97–1.77 (m, 1H), 1.54–1.43 (m, 2H), 1.26 (td, J = 7.1, 5.5 Hz, 6H); 13C NMR (101 MHz, CDCl3) δ 169.47, 167.89, 117.38 (tt, J = 247.3, 29.4 Hz), 110.11 (tt, J = 249.4, 40.3 Hz), 61.87, 61.85, 32.99, 29.12 (t, J = 22.4 Hz), 20.25, 19.62 (t, J = 5.5 Hz), 14.15, 14.09; 19F NMR (376 MHz, CDCl3) δ –116.19 (t, J = 17.2 Hz), –136.03 (dd, J = 53.7, 26.3 Hz); HRMS (ESI+) m/z calcd for C12H17F4O4 [M+H]+: 301.1063, found 301.1062.
Reduction of (5,5,6,6-tetrafluoro-3-iodohexyl)benzene (3b). (5,5,6,6-Tetrafluorohexyl)benzene (8): Compound 3b (150 mg, 0.42 mmol) was dissolved in dry THF (2 mL) and cooled to 0 °C. LiAlH4 (30 mg, 0.79 mmol) was then added and the reaction mixture was allowed to warm to rt and stirred for 16 h (GC-MS control). Saturated solution of NH4Cl (2 mL) and water (30 mL) were added and the reaction mixture was extracted with Et2O. The organic phase was dried (MgSO4) and then concentrated under reduced pressure. The residue was purified by silica gel column chromatography in hexane to afford pure 8 (75 mg of colorless oil, 77% yield). 1H NMR (400 MHz, CDCl3) δ 7.34–7.29 (m, 2H), 7.25–7.18 (m, 3H), 5.71 (tt, J = 54.1, 2.8 Hz, 1H), 2.67 (t, J = 7.5 Hz, 2H), 2.10–1.91 (m, 2H), 1.78–1.69 (m, 2H), 1.69–1.60 (m, 2H); 13C NMR (101 MHz, CDCl3) δ 141.99, 128.53, 128.50, 126.04, 118.13 (tt, J = 245.8, 29.2 Hz), 110.47 (tt, J = 249.1, 41.5 Hz), 35.73, 31.22, 29.78 (t, J = 22.5 Hz), 20.20 (t, J = 3.8 Hz); 19F NMR (376 MHz, CDCl3) δ –116.11 (td, J = 18.2, 2.0 Hz), –135.45 (d, J = 54.0 Hz); HRMS (EI+) m/z calcd for C12H14F4 [M]+: 234.1032, found 234.1033.
Acknowledgements: This work was financially supported by the Academy of Sciences of the Czech Republic (RVO: 61388963). We are grateful to P&M Invest Ltd. for the generous gift of HCF2CF2I.
References
- Dolbier, W. R. Chem. Rev. 1996, 96, 1557.
- Studer, A. Angew. Chem., Int. Ed. 2012, 51, 8950.
- Ji, Y.; Brueckl, T.; Baxter, R. D.; Fujiwara, Y.; Seiple, I. B.; Su, S.; Blackmond, D. G.; Baran, P. S. Proc. Natl. Acad. Sci. USA 2011, 108, 14411.
- Wei-Yuan, H.; Yuan, X. Chin. J. Chem. 1990, 8, 362.
- Fujiwara, Y.; Dixon, J. A.; O'Hara, F.; Funder, E. D.; Dixon, D. D.; Rodriguez, R. A.; Baxter, R. D.; Herle, B.; Sach, N.; Collins, M. R.; Ishihara, Y.; Baran, P. S. Nature 2012, 492, 95.
- Zhang, C. Adv. Synth. Cat. 2014, 356, 2895.
- Nagib, D. A.; MacMillan, D. W. Nature 2011, 480, 224.
- Greiner, J.; Milius, A.; Riess, J. G. J. Fluorine Chem. 1992, 56, 285.
- Takeyama, Y.; Ichinose, Y.; Oshima, K.; Utimoto, K. Tetrahedron Lett. 1989, 30, 3159.
- Bravo, A.; Bjørsvik, H.-R.; Fontana, F.; Liguori, L.; Mele, A.; Minisci, F. J. Org. Chem. 1997, 62, 7128.
- Chen, Q.-Y.; Qiu, Z.-M.; Yang, Z.-Y. J. Fluorine Chem. 1987, 36, 149.
- Beier, P.; O’Hagan, D.; Pearson, C.; Petty, M. C.; Slawin, A. M. Z. J. Fluorine Chem. 2005, 126, 671.
- Zhang, C. P.; Chen, Q. Y.; Guo, Y.; Xiao, J. C.; Gu, Y. C. Chem. Soc. Rev. 2012, 41, 4536.
- Huang, W.-Y.; Zhang, H.-Z. J. Fluorine Chem. 1990, 50, 133.
- Habib, M. H.; Mallouk, T. E. J. Fluorine Chem. 1991, 53, 53.
- Qiu, Z.-M.; Burton, D. J. J. Org. Chem. 1995, 60, 3465.
- Mizuta, S.; Verhoog, S.; Engle, K. M.; Khotavivattana, T.; O'Duill, M.; Wheelhouse, K.; Rassias, G.; Medebielle, M.; Gouverneur, V. J. Am. Chem. Soc. 2013, 135, 2505.
- Iqbal, N.; Jung, J.; Park, S.; Cho, E. J. Angew. Chem., Int. Ed. Engl. 2014, 53, 539.
- Wozniak, L.; Murphy, J. J.; Melchiorre, P. J. Am. Chem. Soc. 2015, 137, 5678.
- Wang, Y.; Wang, J.; Li, G. X.; He, G.; Chen, G. Org. Lett. 2017, 19, 1442.
- Chernykh, Y.; Hlat-Glembová, K.; Klepetářová, B.; Beier, P. Eur. J. Org. Chem. 2011, 2011, 4528.
- Beier, P.; Chernykh, Y.; Opekar, S.; Klepetářová, B. Synlett 2012, 23, 1187.
- Chernykh, Y.; Beier, P. J. Fluorine Chem. 2013, 156, 307.
- Chernykh, Y.; Jurásek, B.; Beier, P. J. Fluorine Chem. 2015, 171, 162.
- Václavík, J.; Chernykh, Y.; Jurásek, B.; Beier, P. J. Fluorine Chem. 2015, 169, 24.
- Li, L.; Ni, C.; Xie, Q.; Hu, M.; Wang, F.; Hu, J. Angew. Chem., Int. Ed. Engl. 2017, 56, 9971.
- Yoshioka, E.; Kohtani, S.; Jichu, T.; Fukazawa, T.; Nagai, T.; Kawashima, A.; Takemoto, Y.; Miyabe, H. J. Org. Chem. 2016, 81, 7217.
- Tiwari, D. P.; Dabral, S.; Wen, J.; Wiesenthal, J.; Terhorst, S.; Bolm, C. Org. Lett. 2017, 19, 4295.
Recommended for publication by Prof. Sergey Igumnov
Fluorine Notes, 2017, 115, 3-4
