Received: September 2018
DOI 10.17677/fn20714807.2018.05.03
Fluorine Notes, 2018, 120, 5-6
Synthesis of some 1,3-diketones of thiophene type, which contain perfluorinated linear substituents
I.V.Taidakov, Yu. A. Kreshchenova, Е.А. Dolotova
P.N. Lebedev Physical Institute of the Russian Academy of Sciences, 119991, Leninskiy Prospekt, 53, Moscow, Russia
e-mail: taidakov@gmail.com
Abstract: A simple preparative method for obtainment of 1,3-diketones containing 2-thienyl and С5-С8 perfluorinated linear substituents, by using condensation of 2-acetylthiophen and corresponding perfluorinated acids esters in the presence of NaH in tetrahydrofuran media, was developed. Reaction optimal conditions, which provide high products yields, were detected.
Keywords: perfluorinated 1,3-diketones, thiophen, Claisen condensation.
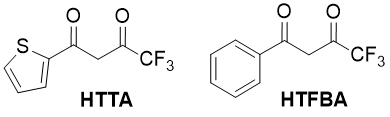
Fatty-aromatic 1,3-diketones, containing perfluorinated substituents, are important starting compounds in the synthesis of d- f-elements coordination compounds. Such compounds have many practical properties (capacity for luminescence, volatility under vacuum, solubility in organic solvents) [1]. They are widely applied as extragents during the nuclear cycle products and rare earth elements separation process [2,3], in analytical chemistry [4], in organic synthesis [5]. Some of these compounds, such as 2-thenoyltrifluoroacetone (НТТА, 4,4,4-trifluoro-1-(2-thienyl)-butanedione-1,3), and lesser, trifluorobenzoylacetone (HTFBA, 4,4,4-trifluoro-1-phenyl-butanedione-1,3), are large-tonnage products and methods of their synthesis are well detailed [6].
Picture 1.
Compounds, that have perfluorinated substituents of mid-length (3-10 carbons in a linear chain) are of interest for the investigation purposes. It was shown that perfluorinated substituent’s moderate elongation in 1,3-diketones of pirazol range leads to strengthening of luminescence intensity of europium complex compound with such diketones, at the same time crystallinity degree of the films, obtained from these compounds, and their solubility in organic solvents changes [7]. The given factors play an important role when new materials for organic light-emitting diodes active layers (OLED – organic light-emitting diode) and other optoelectronic devices are being searched [8].
As a part of a search for new effective luminophores on base of coordinative compounds of lantanides it was of interest to synthesize analogues of a well-known ligand НТТА, which has heavier perfluorinated substituents.
Claisen condensation of 2-acetylthiophen with perfluorinated acids esters is the main synthetic method for such diketones. For 2-thenoyltrifluoroacetone itself there are several dozen variants of carrying out the condensation, different choice of bases, solvents and reaction conditions described in literature, the more suprising is the fact that for diketones with С2F5 [9] and С3F7 radicals [10], only one way of the reaction is described– with NaOMe as a base in diethyl ether media. Diketones which contain С4-С8 substituents up to the present moment were not described at all.
Scheme 1.
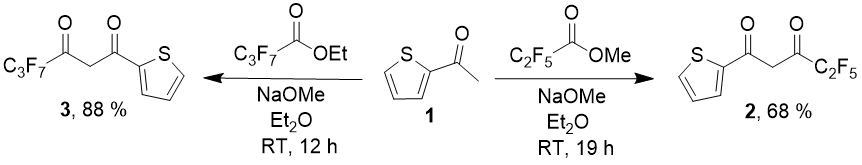
In both cases there is needed a difficult procedure of product isolation, that includes steam distillation, precipitation of cuprous (for compound 2) or magnesiun (for compound 3) chelate, its acid decomposition and further vacuum distillation.
We suggested a method for condensation of methyl or ethyl perfluorinated carbonic acids esters in the presence of sodium hydride as a base in anhydrous tetrahydrofuran. Product of the reaction is separated by one-stage vacuum distillation.
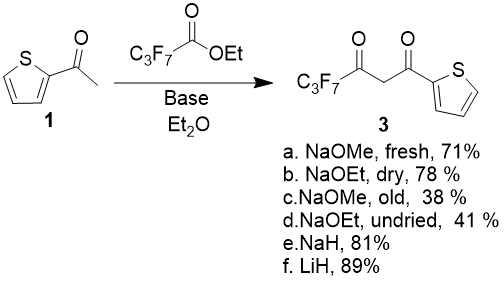
Different bases, first of all, sodium ethoxide and sodium methoxide were tested, but it was found out that product’s yields heavily varies depending on alcoholate’s quality.
Condensation of 2-acetylthiophen with methyl 2,2,3,3,4,4,4-heptafluorobutanoate (as a model compound) in diethyl ether media give the maximum product yields 71 and 79 % when using commercial dry NaOMe from freshly opened tare and freshly prepared NaOEt, previously dried under vacuum 0,1 torr at 60 °С during 6 hours. The same sodium methoxide from tare, which was opened 6 month ago, and sodium ethoxide, obtained by stripping from solution in anhydrous EtOH on rotary evaporator at 60 °С and 10 torr without additional drying gives the condensation products yields - 38 and 41 % correspondingly. Molar ratio of ketone, ester and base in all experiments was 1:1.05:2, initial ketone concentration in solution - 0.2 mol/l, the reaction was held at 5 °С (2 hours), after that it was being additionally refluxed during 5 hours.
So the reaction yields direct dependence from the quality of used alcoholate and difficulty of preparation do not allow to recommend it as an optimal base, at least in case of small amounts of reagent inputs.
Besides sodium alcoholates, there were also tasted NaH (60 % dispersion in mineral oil) and LiH. In the same conditions, using 2 equivalents of hydrides there were obtained 81 and 89 % yields, but the lithium hydride is much more expensive, that is why it does not have substantial advantages. Thus it is necessary to consider NaH as the optimal base. The best yields were obtained when using not less than 2 mol of NaH for 1 mol of ketone. By doing so, total fixation of alcohol, separated during condensation, can be achieved and also errors connected with hydride dispersion instability and partial decomposition while storing are smoothed over. Easy control over the reaction pass by release of hydrogen intensity is an additional advantage of its usage.
Scheme 2.
According to the literature, it is recommended to use different solvents, such as: cyclohexane, toluene, benzene, xylenes, diethyl ether for condensation reactions with sodium hydride [11]. All noted above solvents have the same negative attribute – the reaction mixture stays heterogeneous during all the period, what is more, it’s viscosity grows up as long as diketone sodium derivative is forming. As a rule, this leads to impossibility to mix the reacting mass and as a consequence – to dangerous overheats leading to product yield decrease. The usage of tetrahydrofuran solves this problem because diketones sodium derivatives are easily solved in it, even in the presence of heavy perfluorinated groups in a molecule. Besides that, tetrahydrofuran easily dissolves both 2-acetylthuophen and perfluoroacid ester, although they may not always be mixed with each other without a solvent. As can be seen from above, as far as reaction flows, the reacting mixture becomes less viscous and the problem of heat removal disfunction does not arise.
Regime and order of reagents mixing is an essential condition for achievement of diketones high yields. We found out that optimal is both components solution dropwise addition to suspension of NaH in tetrahydrofuran and that the mixture temperature must not be higher than + 10 °С. For inputs about 100 mmol of initials, it takes 4-6 hours for reaction to be finished. Sometimes recommended in the literature, reaction mass refluxing is not needed, moreover in the given conditions it decreases the product yield on 20-25 % due to gumming.
Scheme 3.
It should be noted, that condensation reaction, when using alkali metals hydrides, is autocatalytic (it speeds up as far as sodium alcoholate accumulates in the mixture), what can lead to hazard effect [12]. If some amount of absolute EtOH (about 0,2-0,5 ml for 100 mmol of hydride) is added into hydride suspension before the reaction is started, then this eliminates the problem and allows synthesis to flow smoothly.
Due to the fact that when the reaction is finished, mixture can contain some amount of unreacted NaH, it is efficient to add absolute ethanol in amount that is equivalent to the amount of inputted NaH before decomposition. After that HCl aqueous solution can be added till weak acid medium safely. In order to rise the product yield, avoidance of mass overheat is needed and all reactions should be held at the temperature, not higher than + 10 °С.
Product isolation is held as usual, at that, due to perfluorinated diketones high acidity, alkali washings should be avoided. Raw diketone vacuum distillation (0,5-1 torr) is the best way for its purification, at that, even heavy diketones can be distilled without decomposition, because the presence of long perfluorinated substituents significantly decreases boiling temperature and raises volatility in comparison to its aliphatic analogues.
Synthesized compounds are light-yellow or colorless crystal materials. Melting and boiling temperatures consistently grow up alongside with elongation of the chain. All of them were characterized by the number of physicochemical methods, its content and purity were also proved by the elemental analysis results.
According to 1Н NMR data, all three diketones in solutions in CDCl3 exist only in enol form. This is proved by broad singlets at 14,9 -15,3 ppm which are relevant to enol form of OH-group proton resonance capture, and also there is only one tight singlet at 6,5 ppm corresponding to CH-group protons signal at enol double bond. Signals that could be referred to protons of СН2-group of ketone form resonance absorption were not found [13]. 19F NMR spectra fully conform to inferred structures.
On the contrary, 13С NMR spectra are less informative, although there is a strong signal at 180-185 ppm that corresponds to quaternary carbon atom of carbonyl group, three signals in the field 136,133, 129 ppm, which should be referred to СН-groups carbon atoms of thiophen ring, and signal at 95-97 ppm, which is referred to СН-group at enol multiple bond. Other signals could not be divided and identified, but they also prove the presence of only one (enol) tautomer in the solution [14].
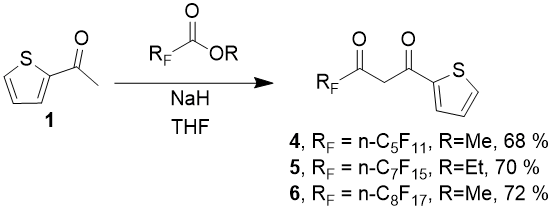
As can be seen from the above, a preparative method for obtainment of thiophen range diketones, which contain perfluorinated substituents of medium length, was developed, and three new representatives of these compounds: 4,4,5,5,6,6,7,7,8,8,8 -undecafluoro-1-(2-thienyl)-octadione-1,3 (4), 4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-pentadecafluoro-1-(2- thienyl)-decanedione-1,3 (5) and 4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoro-1-(2-thienyl)-undecanedione-1,3 (6) with high yields were obtained for the first time.
Experimental
Tetrahydrofuran (p.a., Component-Reactive, Russia) is distilled over sodium metal in the presence of benzophenone and kept in argon atmosphere. Methyl or ethyl perfluorinated acids ethers (SIA «P&M-Invest», Russia) are being held over molecule sieves 4A during 7 days. Other reagents, produced by Acros Organics (Belgium) are used without additional purification. Sodium hydride was used as a commercial 54-60 % suspension in paraffinic oil. NaH exact content is not identified.
1H, 19F and 13С NMR spectra were recorded on «Bruker AM-300» at 300, 282 and 151 MHz correspondingly, at 25 °С for solutions in CDCl3. Chemical shifts for 1H spectra are given relatively to solvent residual signal (δ 7,26) and are given in ppm relatively to TMS. 19F spectra chemical shifts are given in ppm relatively to CFCl3. Downfield shifts are positive. Elemental analysis was held in the INEOS RAS Microanalysis laboratory.
The general methodology for 1,3-diketones preparation.
To NaН suspension 2,0 g. ( 50 mmol, 60 % dispersion) in 100 ml of anhydrous tetrahydrofuran under Ar atmosphere, while intensive stirring by magnetic mixer, 0,2 ml of absolute EtOH was carefully added, then the solution was cooled in ice bath till 0÷5°С, and the mixture of 3,2 g (25 mmol) 2-acetylthiophen and 25 mmol of corresponding methyl or ethyl perfluorocarboxylic acid ether dissolved in 40 ml of tetrahydrofuran was added by drops slowly (2 hours) at this temperature. Reaction mixture gradually becomes pink and then brown. Addition was regulated so that the release of hydrogen was not too rapid. After completion, the mixture was being stirred for 6 hours more, at 20÷25°С or was left for night. After this, the mixture was cooled to 0÷5°С, then 5 ml of absolute EtOH was added by drops and stirred during 20 min. The solution was put in separatory funnel containing mixture of 100 g of processed ice and 10 ml of concentrated НСl and was extracted by 3*50 ml of ethyl acetate. Combined organic extracts were washed with 50 ml of NaCl sole solution, dried by MgSO4 and are vaporized in vacuum. The residue was distilled from Claisen flask without fractioning column at 0,5 torr.
4,4,5,5,6,6,7,7,8,8,8 -undecafluoro-1-(2-thienyl)-octadione-1,3 (4)
Light-yellow crystalline mass. Yield – 7,2 g (68 %), boiling temp. 92÷95°С/0,5 torr. Found (%):C, 34,19; H, 1,22; F, 49,57. C12H5F11O2S. Calculated (%):C, 34,14; H, 1,19; F, 49,50. NMR 1H δ: 15,3 (broad s., 1H, OH), 7,88 (d, 1Н, J = 3,8 Hz, СHTh), 7,80 (d, 1Н, J = 4,8 Hz, СHTh), 7.23 (t, 1Н, J = 10,1 Hz, СHTh), 6,51 (s, 1Н, СH=); NMR 19F δ: -81,61 (s, 3F, CF3), -121,23 (s, 2F, CF2), -123.31 (s, 4F, CF2),-126.97 (s, 2F, CF2).
4,4,5,5,6,6,7,7,8,8,9,9,10,10,10-pentadecafluoro-1-(2-thienyl) -decanedione-1,3 (5)
Colorless crystalline mass. Yield – 9,3 g (70 %), boiling temp. 122÷124°С/0,5 torr. Found (%):C, 32,26; H, 1,05; F, 54,61. C14H5F15O2S. Calculated (%):C, 32,20; H, 0,97; F, 54,57. NMR 1H δ: 14,9 (broad s., 1H, OH), 7,88 (d, 1Н, J = 3,7 Hz, СHTh), 7,79 (d, 1Н, J = 4,9 Hz, СHTh), 7.23 (t, 1Н, J = 4,5 Hz, СHTh), 6,50 (s, 1Н, СH=); NMR 19F δ: -81,65 (s, 3F, CF3), -121,16 (t, 2F, J = 11,3 Hz, CF2), -122.30 (s, 2F, CF2), -122.76 (s, 2F, CF2), -123.10 (s, 2F, CF2) -123.51(s, 2F, CF2), -126.91 (s, 2F, CF2).
4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoro-1-(2-thienyl)- undecadione-1,3 (6)
Colorless crystalline mass. Yield – 10,2 g (72 %), boiling temp. 142÷145°С/0,5 torr. Found (%):C, 31,54; H, 0,91; F, 56,58. C15H5F17O2S. Calculated (%): C, 31,48; H, 0,88; F, 56,44. NMR 1H δ: 15,0 (broad s., 1H, OH), 7,88 (d, 1Н, J = 3,7 Hz, СHTh), 7,79 (d, 1Н, J = 4,8 Hz, СHTh), 7.23 (t, 1Н, J = 4,4 Hz, СHTh), 6,51 (s, 1Н, СH=);19F δ: -81,66 (s, 3F, CF3), -121,16 (t, 2F, J = 11,3 Hz, CF2), -122.26 (s, 2F, CF2), -122.64 (s, 4F, CF2), -123.09 (s, 2F, CF2) -123.50(s, 2F, CF2), -126.92 (s, 2F, CF2).
Acknowledgements
The authors are thankful to SIA «P&M-Invest» for perfluorinated acids ethers samples (ethyl perfluorononanoat, cat. № 0385, methyl perfluorohexanoat, cat. № 1282 and methyl perfluoroctanoat, cat. № 1406). This work was partly supported by the Russian Science Foundation (RSF) grant No 17-72-20088 and the Russian Foundation of Fundamental Research (grant No 18-02-00653 a).
Literature
- Compr. Coord. Chem. II V.1, 2nd Ed., J. A. McCleverty, T.J. Meyer, 2003 Elsevier Ltd., N.Y., 2003, p. 97.
- A.-S. Chauvin, F. Gumy, I. Matsubayashi, Y. Hasegawa, J.-C. G. Bünzli, Eur. J. Inorg. Chem. 2006, 473–480.
- V. Babain, V. Romanovskii, V. Starchenko, A. Shadrin, G. Kudinov, S. Podoinitsyn, Y. Revenko, J. Nuclear Sci. and Techn., 2002, 39, 267-269.
- Е. Nakamura, Y.Hiruta, T. Watanabe, N. Iwasawa, D.Citterio, K. Suzuki, Anal. Sci., 2015, 31(9), 923-928.
- A. Kel’in, A. Maioli, Current Org. Chem.,2003, 7(18), 1855-1886.
- V. G. Isakova, T. S. Khlebnikova, F. A. Lakhvich, Russ. Chem. Rev., 2010, 79(10), 849 – 879.
- E.A. Varaksina, I.V. Taydakov, S.A. Ambrozevich, A.S. Selyukov, K.A. Lyssenko, L.T. Jesus, R.O. Freire, J. of Lumin., 2018, 196, 161–168.
- I.V.Taydakov, A. A. Akkuzina, R. I. Avetisov, A. V. Khomyakov, R. R. Saifutyarov, I. Ch. Avetissov, J. of Lumin., 2016, 177, 31-39.
- R.A. Moore, R. Levine, J. Org. Chem., 1964, 29, 1439 – 1444.
- L. B. Barkley, R. Levine, JACS, 1951, 73, 4625-4627.
- Organic reactions,V.1, R. Adams, John Wiley & Sons, Inc., N.Y., 1942, p.266.
- Advanced Organic Chemistry, 6th Ed., J. March, M.B. Smith, John Wiley & Sons, Inc., Hoboken, 2007, p.1455.
- A. G. Cook, P. M. Feltman, J. Chem. Educ., 2007, 84 (11), 1827-1829.
- M. P. Sammes, P. N. Maini, Magnet. Res. Chem.,1987, 25, 372-374.
Recommended for publication by Prof. S.R. Sterlin
Fluorine Notes, 2018, 120, 5-6
