Received: September 2018
DOI 10.17677/fn20714807.2018.06.01
Fluorine Notes, 2018, 121, 1-2
Synthesis of Chiral Fluorinated Amino Acids by Eosin Y Catalyzed Perfluoroalkyl Radical Addition to Dehydroamino Acids
Tomoko Yajima*, Mako Ikegami
Department of Chemistry, Faculty of Science, Ochanomizu University, Otsuka, Bunkyo-ku, Tokyo 112-8610, Japan
Email: yajima.tomoko@ocha.ac.jp
Abstract: Fluorine-containing amino acids are among the most desirable compounds in the medicinal field. Previously, we reported several methods for the synthesis of β-perfluoroalkyl amino acids based on radical perfluoroalkylation under UV irradiation or In catalyzed conditions. In this paper, we describe the synthesis of β-perfluoroalkyl amino acids by metal-free visible light-induced perfluoroalkylation to dehydroamino acids using Eosin Y as an organic photoredox catalyst. The use of a chiral auxiliary method produced chiral amino acids high stereoselectivity. This ecological method will produce various perfluoroalkylated chiral amino acid derivatives.
Keywords: organic dye; perfluoroalkylation; amino acid; chiral auxiliary; visible light
Fluorine-containing molecules, in particular fluorinated amino acids and their peptides, are among the most promising compounds in the fields of medicinal chemistry and supramolecular sciences [1–4]. Because of the active research in peptide drug discovery [5, 6], the development of specialty peptides is anticipated. Perfluoroalkyl-containing α-amino acids are expected to provide a novel nature to the peptide because of their chemical and thermal stabilities, and other properties such as hydrophobicity, bulkiness, and charge distribution are adjustable by changing the perfluoroalkyl groups. However, the efficient synthesis methods for perfluoroalkyl-containing chiral amino acids are limited [2, 4]. We have reported several synthetic approaches for β-perfluoroalkyl-α-amino acids based on the radical reaction [7–9]. Although our previous methods provided various perfluoroalkylated amino acids, the process imposed limitations, such the use of metal indium or a high-pressure Hg lamp for radical initiation, which had low yield or low selectivity.
In the last decade, visible light-induced photoredox reactions, particularly using organic dye as a catalyst, have been actively researched as a metal-free and sustainable process, which is suitable for pharmaceuticals preparation [10–12]. In a previous study, we developed Eosin Y catalysed iodoperfluoroalkylation of alkenes and alkynes [13]. The reaction proceeded smoothly to give the corresponding iodoperfluoroalkylated product using only 1 mol% of Eosin Y under visible light irradiation for 1 h using various perfluoroalkyl iodides as the fluorine source (Scheme 1). Unfortunately, the reaction substrates were limited to simple terminal alkenes and alkynes. Therefore, this time, we applied the reaction principle to dehydroamino acids for the metal-free synthesis of perfluoroalkylated amino acids. Here, we reported the Eosin Y-catalyzed visible light-induced perfluoroalkylation of dehydroamino acids. A stereoselective reaction was also performed using a chiral auxiliary method.
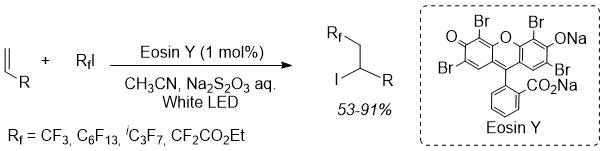
Scheme 1. Our previous photoinduced iodoperfluoroalkylation.
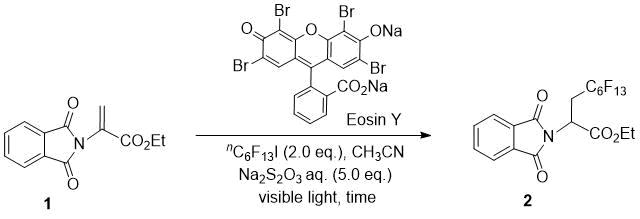
First, we investigated the photoinduced radical addition of perfluorohexyl iodide to N-phthalimide-protected dehydroamino acid 1 to optimize the reaction conditions (Table 1). The reaction of 1 with 2.0 equiv. of perfluorohexyl iodide using 1 mol% Eosin Y as the photocatalyst with irradiation for 1 h by a 12-W white light-emitting diode (LED) lamp in CH3CN and the presence of aqueous Na2S2O3 was performed according to our previous reaction conditions (entry 1) [13]. The corresponding hydroperfluorohexylated product 2 was obtained in 7% yield; however, in our previous report, the reaction with simple olefins gave iodoperfluoroalkylated product even in the same reaction conditions. We then increased the reaction time (entries 2–4); the longer reaction time produced a higher product yield, and with irradiation for 16 h, the yield was increased up to 65%. The use of 5 mol% Eosin Y was equally effective (entry 5). However, the reaction using a green LED lowered the yield (entry 6).
Table 1. Optimization of reaction conditions.
|
Entry |
Eosin Y |
Light source |
Time (h) |
yield |
|
1 |
1 mol% |
12-W White LED |
1 |
7% |
|
2 |
1 mol% |
12-W White LED |
5 |
24% |
|
3 |
1 mol% |
12-W White LED |
8 |
40% |
|
4 |
1 mol% |
12-W White LED |
16 |
65% |
|
5 |
5 mol% |
12-W White LED |
5 |
65% |
|
6 |
1 mol% |
6-W Green LED |
16 |
19% |
Next, the reactions using chiral substrates with various perfluoroalkyl iodides were investigated (Table 2). The reaction between chiral substrate 3 [7], containing both phthalimide and camphorsultam moieties, and a perfluoroalkyl iodide proceeded smoothly to give product 2a in 82% yield as a single stereoisomer under visible light irradiation for 6 h using 1 mol% Eosin Y (entry 1). However, the use of perfluorohexyl chloride instead of perfluorohexyl iodide did not give the desired perfluoroalkylated product. In fact, the reaction using perfluorobutyl or isopropyl iodide gave the corresponding perfluoroalkylated product 2b or 2c in good yield with high diastereoselectivity (entries 2, 3). Meanwhile, the reaction with trifluoromethyl iodide produced a lower yield, but once the reaction time was increased, the yield was improved (entries 4, 5). In our previous report [7], the diastereoselectivity of 2d was lower than that of the other perfluoroalkyl adducts; however, excellent stereoselectivity was obtained under this photoredox process. The reaction using iododifluoroacetate as the fluorine source only produced a 53% yield despite the 16 h reaction time, but the use of 5 mol% Eosin Y improved the product yield to 73% (entries 6, 7). These products could then be easily transformed to amino acids via the removal of the sultam auxiliary and the deprotection of the phthalimide [7].
Table 2. Stereoselective hydroperfluoroalkylation.
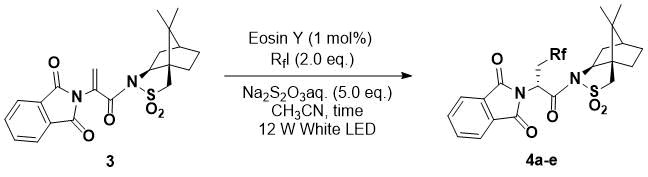
|
Entry |
Rf = |
Time (h) |
Product |
Yield |
% dea |
|
1 |
C6F13 |
6 |
4a |
82% |
>98 |
|
2 |
C4F9 |
6 |
4b |
89% |
>98 |
|
3 |
i-C3F7 |
6 |
4c |
84% |
>98 |
|
4b |
CF3 |
6 |
4d |
44% |
>98 |
|
5c |
CF3 |
16 |
4d |
72% |
>98 |
|
6 |
CF2CO2Et |
16 |
4e |
53% |
>98 |
|
7d |
CF2CO2Et |
16 |
4e |
73% |
>98 |
aDiasteromer ratios were determined by 1H NMR.
b19 eq. of CF3I
was used.
c16 eq. of CF3I was used.
d5 mol% Eosin Y was
used.
To confirm the origin of the hydrogen, a labeling experiment using D2O was performed (Scheme
2). The reaction between 3 and perfluorohexyl iodide using D2O instead
of H2O produced a 66% α-deuterated product in 83% yield.
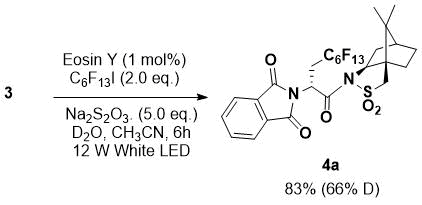
Scheme 2. Labeling experiment.
A possible reaction pathway for the hydroperfluoroalkylation is shown in Scheme 3. The perfluoroalkyl radical was generated by the oxidative quenching cycle of the photo-excited Eosin Y [7]. The perfluoroalkyl radical was added to the dehydroamino acid to give a captodatively stabilized radical intermediate A [8]. Then, the radical intermediate A was oxidized by Na2S2O3 to be the anion B. The final stereoselective protonation of B by D2O afforded the corresponding hydroperfluoroalkylated product. In this reaction, iodine radical transfer is unfavorable because intermediate radical A is stable and sterically crowded compared to the reaction with decene.
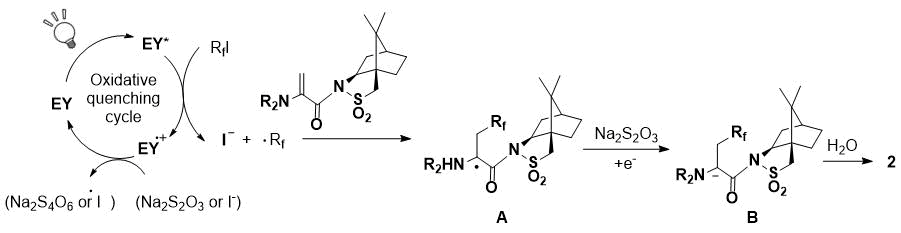
Scheme 3. Proposed mechanism.
We accomplished the Eosin Y-catalyzed visible light-induced hydroperfluoroalkylation of dehydroamino acids using perfluoroalkyl iodides as the fluorine source. The use of camphorsultam as the chiral auxiliary substrate resulted in excellent stereoselectivity, and the reaction was useful for various perfluoroalkyl iodides. The process is new, sustainable, and efficient for the synthesis of chiral perfluoroalkylated amino acids.
Experimental
All reactions were carried out under a nitrogen atmosphere. Visible light irradiations were performed with a 12 W LED lamp (Hayashi watch-works Co., Ltd., SPA-10SW). 1H, 13C and 19F NMR spectra were recorded on a JEOL JNM-ECS400 (399.78 MHz for 1H, 100.5 MHz for 13C, and 376.2 MHz for 19F) or JNM-ECX500 (500.16 MHz for 1H, 125.8 MHz for 13C, and 470.6 MHz for 19F) spectrometer with CDCl3 as the solvent and tetramethylsilane (δ 0 ppm for 1H), chloroform-d (CDCl3: δ 76.9 ppm for 13C) and trichlorofluoromethane (CFCl3 : 0 ppm for 19F) as an internal standard unless otherwise noted. IR spectra were taken on JASCO FTIR-4100. HRMS were obtained with a Thermo Fisher Exactive (ESI, APCI), JEOL JMS-T100GC (FD) or JEOL JMS-700 (FAB). Precoated Merck Kieselgel 60 F254 and Kanto silica gel 60 (spherical neutral) were used for thin layer chromatography and flash chromatography, respectively. All chemicals were used without further purification.
General procedure for visible light induced perfluoroalkylation.
To a two-necked flask containing olefin (0.2 mmol) in CH3CN (5 mL) under a nitrogen atmosphere were added eosin Y-2Na (1.4 mg, 0.002 mmol) solved in water (1 mL), Na2S2O3 (158.0 mg, 1.0 mmol) and perfluoroalkyl iodide (0.4 mmol). The mixture was irradiated with 12 W White LED lamp at room temperature with stirring. After the reaction was completed, the mixture was extracted with diethyl ether, dried over Na2SO4, filtered, and concentrated in vacuo. The crude product was purified by flash chromatography (silica; hexane) to give the product.
Ethyl 2-(1,3-dioxoisoindolin-2-yl)-3-perfluorohexylpropanoate (2)
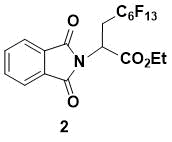
White powder. 1H NMR (400 MHz, CDCl3) 7.90 (m, 2H), 7.78 (m, 2H), 5.32 (dd, J = 10.0, 4.0 Hz, 1H), 4.27 (m, 2H), 3.17 (m, 2H), 1.24 (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3) 167.3, 167.0, 134.5, 131.6, 123.8, 62.9, 45.2, 29.6 (t, JC–F = 20.2 Hz, C6F13C), 14.0; IR (KBr) 2987, 1724, 1718, 1387, 1236, 1072, 860, 848, 720, 530 cm-1; HRMS (ESI-) m/z calcd for C19H11F13NO4 [M-H] - 564.0481, found 564.0479.
This compound has been previously prepared and characterized [7]
N-{(R)-2-(1,3-Dioxoisoindolin-2-yl)-3-perfluorohexylpropanoyl}-(1R,2S,4S)-bornane-10,2-sultam (4a)
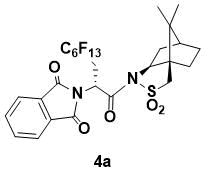
White powder. 1H NMR (400 MHz, CDCl3) 7.86 (m, 2H), 7.73 (m, 2H), 5.67 (dd, J = 9.4, 3.2 Hz, 1H), 4.06 (m, 1H), 3.53 (d, J = 13.6 Hz, 1H), 3.28 (d, J = 13.6 Hz, 1H), 3.21 (m, 1H), 2.75 (m, 1H), 2.10 (m, 2H), 1.87 (m, 3H), 1.36 (m, 2H), 0.88 (s, 6H); 13C NMR (150 MHz, CDCl3) 167.6, 166.6, 134.3, 132.0, 123.5, 67.0, 53.4, 48.4, 47.6, 46.5, 45.1, 37.8, 33.3, 30.7 (t, JC–F = 29.1 Hz, C6F12C), 26.2, 20.5, 19.8; IR (KBr) 1707, 1334, 1318, 1238, 1226, 1190 cm-1;HRMS (ESI+) m/z calcd for
C27H23F13N2O5SNa [M+Na]+ 757.1012, found 757.0995.
This compound has been previously prepared and characterized [7]
N-{(R)-2-(1,3-Dioxoisoindolin-2-yl)-3-perfluorobutylpropanoyl}-(1R,2S,4S)-bornane-10,2-sultam (4b)
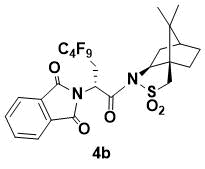
White powder. 1H NMR (400 MHz, CDCl3) 7.86 (m, 2H), 7.74 (m, 2H), 5.67 (dd, J = 9.4, 3.2 Hz, 1H), 4.07 (m, 1H), 3.42 (d, J = 13.6 Hz, 1H), 3.36 (d, J = 13.6 Hz, 1H), 3.23 (m, 1H), 2.76 (m, 1H), 2.14 (m, 1H), 2.05 (m, 1H), 1.88 (m, 3H), 1.38 (m, 2H), 0.88 (s, 6H); 13C NMR (150 MHz, CDCl3) 167.6, 166.6, 134.3, 132.1, 123.6, 67.0, 53.5, 48.5, 47.7, 46.5, 45.2, 37.9, 33.4, 30.6 (t, JC–F = 29.1 Hz, C6F12C), 26.3, 20.5, 19.8; HRMS (ESI+) m/z calcd for C25H24F9N2O5S [M+H]+ 635.1262, found 635.1248.
N-{(R)-2-(1,3-Dioxoisoindolin-2-yl)-3-perfluoroisopropylpropanoyl}-(1R,2S,4S)-bornane-10,2-sultam (4c)
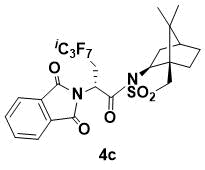
White powder. 1H NMR (400 MHz, CDCl3) 7.84 (m, 2H), 7.75 (m, 2H), 5.67 (m, 1H), 4.06 (dd, J = 7.2, 4.8 Hz, 1H), 3.58 (m, 1H), 3.41 (d, J = 13.6 Hz, 1H), 3.34 (d, J = 13.6 Hz, 1H), 2.50 (m, 1H), 2.14 (m, 2H), 1.88 (m, 3H), 1.38 (m, 2H), 0.87 (s, 3H), 0.85 (s, 3H); 13C NMR (100 MHz, CDCl3) 167.1, 166.4, 134.3, 131.8, 123.5, 66.9, 53.4, 48.4, 47.6, 47.4, 45.1, 37.8, 33.3, 28.8 (d, JC–F = 20.1 Hz, i-C3F7C), 26.2, 20.5, 19.8; HRMS (ESI+) m/z calcd for C24H23F7N2O5SNa [M+Na]+ 607.1108, found 607.1097.
This compound has been previously prepared and characterized [7]
N-{(R)-2-(1,3-Dioxoisoindolin-2-yl)-3-trifluoromethylpropanoyl}-(1R,2S,4S)-bornane-10,2-sultam (4d)
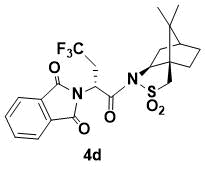
White powder. 1H NMR (400 MHz, CDCl3) 7.85 (m, 2H), 7.76 (m, 2H), 5.55 (dd, J = 10.4, 3.2 Hz, 1H), 4.07 (dd, J = 7.2, 4.8 Hz, 1H), 3.39 (d, J = 13.6 Hz, 1H), 3.35 (d, J = 13.6 Hz, 1H), 3.17 (m, 1H), 2.76 (m, 1H), 2.15 (m, 1H), 2.09 (m, 1H), 1.91 (m, 3H), 1.37 (m, 2H), 0.88 (s, 6H); 13C NMR (100 MHz, CDCl3) 167.6, 166.6, 134.3, 132.0, 123.5, 67.0, 53.4, 48.4, 47.6, 46.4, 45.1, 37.8, 33.3, 30.2, 26.2 (q, JC–F = 29.3 Hz, CF3C), 20.5, 19.8; IR (KBr) 1785, 1699, 1400, 1298, 1207, 1120, 1085, 1039 cm-1; HRMS (ESI+) m/z calcd for C22H23F3N2O5SNa [M+Na]+ 507.1172, found 507.1166.
This compound has been previously prepared and characterized [7]
N-{ethyl (R)-2-(1,3-Dioxoisoindolin-2-yl)-4,4-difluoropentanoyl}-(1R,2S,4S)-bornane-10,2-sultam (4e)
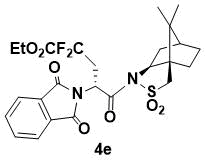
White powder. 1H NMR (400 MHz, CDCl3) 7.83 (m, 2H), 7.71 (m, 2H), 5.59 (dd, J = 10.4, 3.2 Hz, 1H), 4.30 (m, 1H), 4.05 (dd, J = 7.2, 4.8 Hz, 1H), 3.39 (d, J = 13.6 Hz, 1H), 3.33 (d, J = 13.6 Hz, 1H), 3.17 (m, 1H), 2.76 (m, 1H), 2.15 (m, 1H), 2.09 (m, 1H), 1.92 (m, 3H), 1.37 (m, 2H), 1.35 (t, J = 8.0 Hz, 3H), 0.87 (s, 6H); 13C NMR (100 MHz, CDCl3) 167.9, 166.8, 134.2, 132.1, 123.6, 66.3, 53.4, 48.4, 47.7, 47.6, 45.2, 37.9, 33.4, 26.3, 20.6, 19.8; IR (KBr) 1785, 1699, 1400, 1298, 1207, 1120, 1085, 1039 cm-1; HRMS (ESI+) m/z calcd for C25H29F2N2O7S [M+H]+ 539.1663, found 539.1657.
Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Advanced Molecular Transformations by Organocatalysts” (No. 26105717) and Scientific Research (C) (16K05687) from MEXT, Japan. A part of this work was also supported by Koyanagi foundation. We are grateful to P&M Invents for their generous donation of perfluorohexyl chloride and Tosoh F Tech Inc. for their generous donation of trifluoromethyl iodide.
References
- Prakash, R. D. In Organofluorine Compounds in Biology and Medicine, Elsevier, 2015, pp 101−132.
- Uneyama, K. Recent Advances in the Synthesis of Fluorinated Amino Acids, In Fluorine in Medicinal Chemistry and Chemical Biology, Ojima, I., Ed.; Wiley-Blackwell: New York, 2009 pp 213−256.
- Bégué, J.-P.; Bonnet-Delpon, D. In Bioorganic and Medicinal Chemistry of Fluorine, John Wiley & Sons: Hoboken, 2008; pp 146−179.
- Welch, J. T.; Eswarakrishnan, S. In Fluorine in Bioorganic Chemisty, John Wiley & Sons: New York, 1991 pp 7−65.
- Peptide-based Drug Discovery: Challenges and New Therapeutics, Thurston, D., Ed.; Royal Society of Chemistry, 2017.
- Peptide Drug Discovery and Development: Translational Research in Academia and Industry, Castanho, M., Santos N. C. Eds.; Wiley-VCH Verlag, 2011
- Yajima, T.; Yamaguchi, K.; Hirokane, R.; Nogami, E. J. Fluor. Chem., 2013, 150, 1−7.
- Yajima, T.; Tonoi, T.; Nagano, H.; Tomita, Y.; Mikami, K. Eur. J. Org. Chem., 2010, 2461−2464.
- Yajima, T.; Nagano, H. Org. Lett., 2007, 9, 2513−2515.
- Fukuzumi, S.; Ohkubo, K. Org. Biomol. Chem., 2014, 12, 6059-6071.
- Romero, B. A.; Nicewicz, D. Chem. Rev., 2016, 116, 10075-10166.
- Hari, D. P.; König, B. Chem. Comm., 2014, 50, 6688-6699.
- Yajima, T.; Ikegami, M. Eur. J. Org. Chem., 2017, 15, 2126-2129.
- Sun, X.; Wang, W.; Li, Y.; Ma, J.; Yu, S., Org. Lett., 2016, 18, 4638−4641.
- Jiang, H.; He, Y.; Chen,Y.; Yu, S. Org. Lett., 2017, 19, 1240−1243.
- Wang, Y.; Wang, J.; Li, G.-X.; He, G.; Chen, G. Org. Lett., 2017, 19, 1442−1445.
- Tiwari, D. P.; Dabral, S.; Wen, J.; Wiesenthal, J.; Terhorst, S.; Balm, C. Org. Lett., 2017, 19, 4295−4298.
Recommended for publication by Prof. Norio Shibata
Fluorine Notes, 2018, 121, 1-2
