Received: December 2018
DOI 10.17677/fn20714807.2018.06.05
Fluorine Notes, 2018, 121, 9-10
1,1-Dichlorohexafluoroisobutylene and 2,2-Dichloro-3,3-bis(trifluoromethyl)oxirane: Low–Toxic Synthetic Equivalents of Perfluoroisobutylene and Its Derivatives
A.A. Tyutyunovab, A.B. Sin’koab, N.D. Kagramanova, S.R. Sterlina, S.M. Igumnovab
aA.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences,
ul. Vavilova 28, V-334, GSP-1, Moscow, 119991 Russia
bNPO PiM-INVEST LLC, ul. Vavilova 28, Moscow, 119991 Russia
e-mail: tuytuynov@rambler.ru
Abstract: It was demonstrated that the derivatives of α-halo- and α-hydrohexafluoroisobutyric acids and aliphatic compounds containing polyfluorinated tert-butyl groups could be synthesized on the basis of 1,1-dichlorohexafluoroisobutylene and 2,2-dichloro-3,3-bis(trifluoromethyl)oxirane as starting materials.
Keywords: 1,1,1-trichloro-3,3,3-trifluoro-2-(trifluoromethyl)propane-2-ol, 1,1-dichlorohexafluoroisobutylene, 2,2-dichloro-3,3-bis(trifluoromethyl)oxirane, 2-chloro-2-fluoro-3,3-bis(trifluoromethyl)oxirane.
Perfluoroisobutylene (PFIB) is one of the most electrophilic fluoroolefins; it interacts extremely easily with nucleophilic reagents with formation of addition or substitution products [1–2]. In particular, interaction of KF or CsF with PFIB results in ready formation of the perfluoro-tert-butyl anion; its reactions are discussed in detail in reviews [3–4]. It should be also noted that perfluoroisobutylene is the starting compound for synthesis of such products as bis(trifluoromethyl)ketene, thioketene [5–7], and derivatives of perfluoromethacrylic acid [8] that are of apparent synthetic interest.
The main source of PFIB is the C4 hydrocarbon fraction obtained in synthesis of tetrafluoroethylene and hexafluoropropylene by pyrolysis of Freon 22. However, improvement of the pyrolysis technology consisting in performing the process in the presence of water vapor allows eliminating almost completely the formation of the C4 fraction (PFIB, perfluorobutylene-2 and perfluorocyclobutane) [9]. This is an important change as a matter of occupational safety, as PFIB is extremely toxic. Simultaneously, it offers the challenge of searching for safe preparative methods of synthesizing PFIB–based compounds.
As shown earlier, 1,1-dichlorohexafluoroisobutylene (1) reacts in some cases similarly to PFIB and is also a considerably less toxic compound than perfluoroisobutylene (LCt50>25000 and 880 mg.min.m–3, respectively) [10]. Therefore, it is much more convenient to use in practice sufficiently high–boiling and relatively harmless olefin 1 (b.p. 74°C) instead of gaseous PFIB (b.p. 6°C) [11].
The aim of this work was to develop methods of synthesis of compounds containing a perfluoro-tert-butyl group and also derivatives of hexafluoroisobutyric acid without using PFIB. The basic compound for such a study could be 1,1,1-trichloro-3,3,3-trifluoro-2-(trifluoromethyl)propane-2-ol (2), a compound containing a perhalogenated tert-butyl group. Simple methods of synthesizing this compound from hexafluoroacetone and trichloracetic acid or sodium trichloroacetate were developed earlier [12–13].
It is known that tertiary alcohol 2 is deoxychlorinated in a reaction with PCl5 yielding 1,1-dichlorohexafluoroisobutylene (1), but the yield of 1 after 8 h of boiling the reaction mixture does not exceed 42% (in case of 40% conversion of 2) [14]. In view of the fact that vicinal halohydrins are easily reduced to olefins by zinc in alcohol or acetic acid [15–16], we studied the possibility of reducing 2 to 1 as an alternative way of synthesis of dichloroisobutylene 1.
It turned out that interaction of 2 with zinc dust in acetic acid or sulfolane results in reduction of the trichloromethyl group and formation of a mixture of the corresponding tertiary alcohols. Here, when this reaction is carried out in water in the presence of ammonium chloride, it allows selectively synthesizing dichlorohexafluoro-tert-butanol 3.
Scheme 1
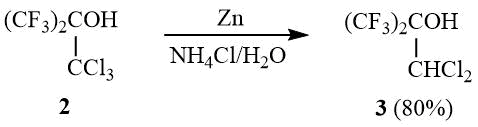
Transformation of the OH group in 2 to an ester group with enhanced nucleofugality, e.g., CH3CO2, CF3CO2, ClCH2CFClCO2, or CH3SO3, allows converting esters 4a-c in a reaction with zinc to dichloroisobutylene 1 with the preparative yield of 60–80%:
Scheme 2
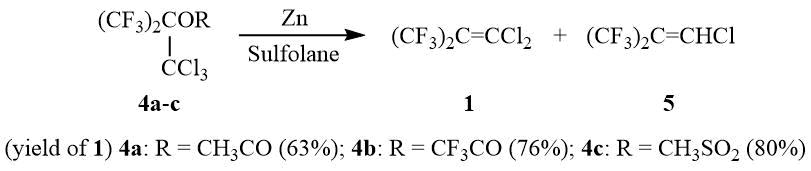
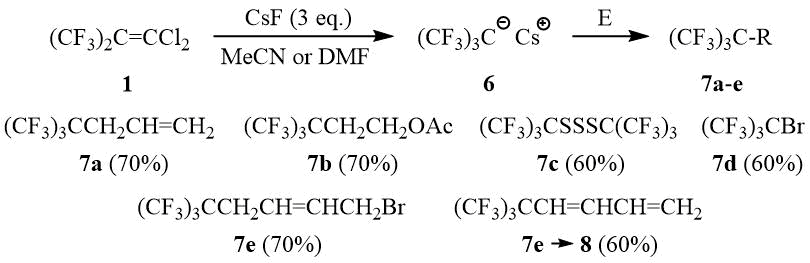
As opposed to PFIB, no formation of perfluoro-tert-butyl anion 6 from dichloroisobutylene 1 in a reaction with stoichiometric amount of KF is observed in such solvents as diglyme, acetonitrile, and DMF even after heating or in the presence of 18-crown-6, Ph4PBr, and (Et2N)3CCl. Also, dichloroisobutylene 1 does not form 6 with CsF in diglyme. However, interaction of 1 with three equivalents of CsF in acetonitrile or DMF leads to formation of 6 (19F NMR δ: –47 (br.s. (CF3)3C–)) that can be used further in a reaction with different electrophilic reagents for synthesis of compounds containing a perfluoro-tert-butyl group, e.g., 7a-e, and also monomer 8.
Scheme 3
At the same time, the reaction of olefin 1 with MeOH in the presence of KF or KOH leads to formation of chlorohexafluoroisobutenylmethyl ether 9 as the main reaction product as a result of nucleophilic addition of methanol with the further dehydrochlorination. Earlier, a similar reaction of 1 with aryl thiols yielded arylchlorohexafluoroisobutenyl sulfides [11].
Scheme 4
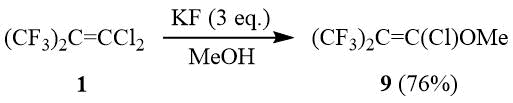
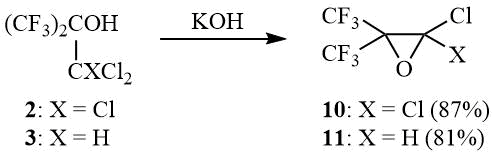
Another important aspect of the chemistry of vicinal haloalcohols, including 1,1,1-trichloro-3,3,3-trifluoro-2-(trifluoromethyl)propane-2-ol (2), is their ability to undergo dehydrohalogenation in a reaction with bases with formation of epoxides, including fluorinated ones [17–18].
Thus, as shown earlier, 2 is dehydrochlorinated by aqueous alkali with formation of 1,1-dichlorohexafluoroisobutylene oxide (10) in 50% yield [12]. As found in the course of the work, reaction 2 with solid KOH allows enhancing the yield of oxide 10 to 87%. Thus, tert-butanol 3 was converted to oxide 11 in 81% yield:
Scheme 5
At the same time, chlorooctafluoro-tert-butanol (CF3)2C(OH)CF2Cl (12) does not undergo dehydrochlorination under exposure to KOH even under prolonged boiling.
High basicity of the fluoride ion [19–20] implied that the reaction of tert-butanol 2 with KF in an aprotic medium would also result in formation of oxide 10. Indeed, alcohol 2 reacted with KF in diglyme at 100÷140°C, but the main reaction product was perfluoroisobutyroyl fluoride (13) obtained in ~50% yield.
Scheme 6
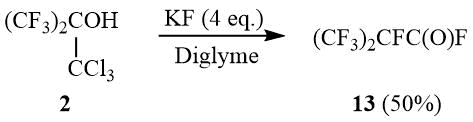
Acylfluoride 13 is obviously formed in the Jocic–Reeve reaction [21–22] with intermediate gem-dichloroepoxide 10 opened by the fluoride anion with formation of perfluoroisobutyroyl chloride and the further fluorination of the chloroformyl group.
The further study of the reaction of oxide 10 with potassium fluoride taken in catalytic amounts showed that oxirane 10 is isomerized in such aprotic solvents as diglyme, acetonitrile, or sulfolane with formation of α-chloroperfluoroisobutyric acid chloride (14).
Scheme 7
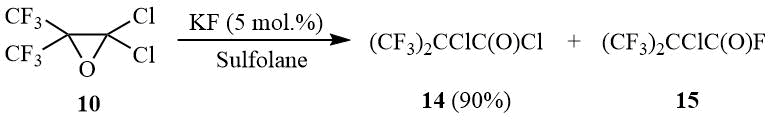
An increase in the amount of KF results in formation of α-chloroperfluoroisobutyroyl fluoride (15) as the main reaction product. Isobutyroyl halides 14–15 are convenient initial compounds for synthesis of bis(trifluoromethyl)ketene [23].
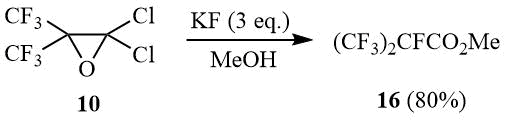
In its turn, interaction of oxide 10 with an excess of potassium fluoride in methanol leads to selective formation of methyl ether of perfluoroisobutyric acid (16).
Scheme 8
Obviously, formation of α-halohexafluoroisobutyl halides 13–15 is a result of a nucleophilic attack at the tertiary carbon atom in oxide 10 by the halide anion (similarly to the opening of the perfluoroisobutylene oxide [24–25]). In the case when the solution contains a catalytic amount of KF, e.g., in such solvents as diglyme or sulfolane, oxide 10 is predominantly isomerized with formation of α-chloroperfluoroisobutyric acid chloride (14). In a methanol solution, in which the solubility of KF is sufficiently high, oxide 10 is opened by the fluoride anion and 16 is formed.
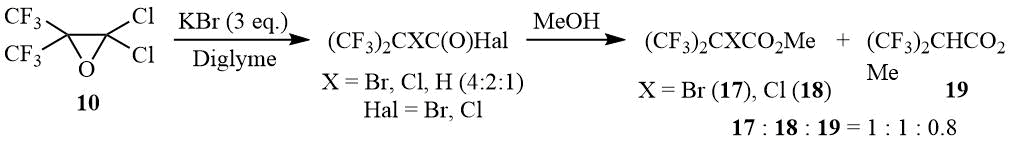
Under exposure to an excess of KBr in diglyme, oxide 10 is also opened with formation of α-bromo-, α-chloro-, and α-hydrohexafluoroisobutyric acid halides in the molar ratio of 4:2:1. Obviously, formation of α-chloroperfluoroisobutyroyl halides is a result of a competitive attack on oxirane 10 by the chloride anion, while α-hydroperfluoroisobutyric acid halides are in all probability formed in the course of a halophilic attack at the tertiary bromine atom. This assumption is confirmed by appearance of bromine as a result of the reaction mixture esterification by methanol and a drastic change in the relative content of α-bromo- and α-hydrohexafluoroisobutyrates (the molar ratio of esters is 17:18:19 = 1:1:0.8), which is most probably related to the higher solubility of potassium halides in the MeOH/diglyme mixture.
Scheme 9
It is of interest to point out that interaction of 10 with the excess of triethylamine in methanol with the further acidification of the reaction mixture yields 2-chloro-1,1,1,3,3,3-hexafluoropropane (20). Its formation can be explained by decarboxylation of the intermediate α-chlorohexafluoroisobutyric acid.
Scheme 10
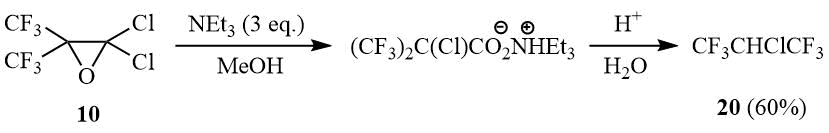
As shown earlier, perfluoroisobutylene oxide isomerizes in a reaction with SbF5 with formation of perfluoroisobutyroyl fluoride [26]; meanwhile, a similar reaction in the presence of HF results in formation of perfluoro-tert-butanol [27]. In its turn, carbinol 2 can be converted in a Swarts reaction to dichlorofluoro-, chlorodifluoro-, and perfluoro-tert-butanol [12, 28–29]. It was of interest to study interaction of oxide 10 with antimony pentafluoride.
It turned out that the reaction of oxirane 10 with SbF5 (0.5 eq.) at 20÷25°C results in formation of a viscous reaction mass that yields, after stirring for 1 h and adding water, a mixture of reaction products containing 25% of oxirane 21 and 63% chlorooctafluoro-tert-butanol (12). Formation of carbinol 12 allows assuming that its intermediate is (CF3)2C(OSbF3Cl)CF2Cl. At the same time, the heating of the reaction mixture with the same reagent ratio with simultaneous distillation of volatile reaction products results in formation of oxirane 21 in 80% yield:
Scheme 11
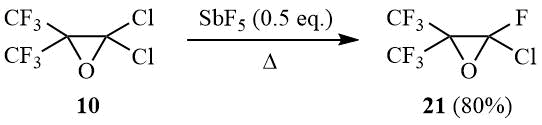
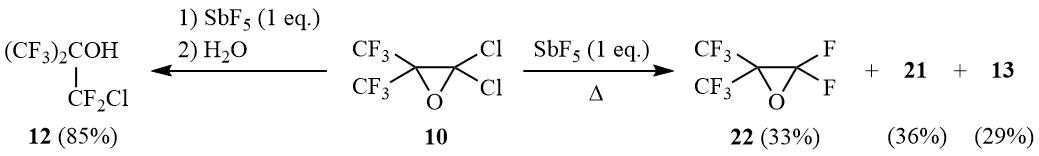
Interaction of equimolar amounts of oxirane 10 and SbF5 under similar conditions (20÷25°C, 1 h of stirring with the further reaction mixture decomposition with water) yields carbinol 12 in 85% yield. The gradual heating of this reaction mixture up to 180÷190°C with simultaneous distillation of volatile reaction products results in formation of a mixture of approximately similar amounts of perfluoroisobutylene oxide (22), oxide 21, and perfluoroisobutyryl fluoride 13:
Scheme 12
Thus, it is shown that dichlorohexafluoroisobutylene 1 and its oxide 10 are of apparent interest as starting compounds for synthesis of substances containing polyfluorinated isopropyl or tert-butyl groups. In particular, dichlorohexafluoroisobutylene 1, being a synthetic equivalent of highly toxic perfluoroisobutylene, can be used for introduction of perfluoro-tert-butyl group into various classes of organic compounds and oxide 10 can be a basis for synthesis of derivatives of α-halo- and α-hydrohexafluoroisobutyric acids.
Experimental
1H, 19F NMR spectra were recorded using a Bruker AVANCE-300 spectrometer at 300 and 282 MHz, accordingly; the external standard was CDCl3. Chemical shifts for 1H spectra are provided vs. the residual signal of the solvent (CHCl3 δ: 7.26) and are provided in ppm vs. TMS. Chemical shifts in 19F spectra are given in ppm vs. CFCl3. Downfield shifts are positive. Mass spectra are recorded using a Finnigan Polaris Q mass spectrometer (Trace GC ultra). Elemental analysis was carried out in Laboratory of Microanalysis of A.N. Nesmeyanov Institute of Organoelement Compounds of Russian Academy of Sciences.
1,1,1-Trichloro-3,3,3-trifluoro-2-(trifluoromethyl)propane-2-ol (2).
Obtained according to the earlier described methods from hexafluoroacetone and CCl3CO2Na [13] or using CCl3CO2SiMe3 according to the methods described further:
Dry DMF (3.5 l) is loaded into 6 l flask equipped with mechanical stirrer, thermometer and reflux condenser (–78°C), connected with Tishchenko flask with conc. H2SO4, then 500 g (8.6 mol) of dry KF are added under stirring, 1000 g (6 mol) of hexafluoroacetone is fed into suspension obtained at 15÷20°C, 1550 g (6.58 mol) of trimethylsilyl trichloroacetate are added dropwise at 5÷10оС, the reaction mixture is warmed up to ~25оС, stirred for 4–5 h, left overnight, 516 g (2.19 mol) of trimethylsilyl trichloroacetate is added gradually at 15÷20°C, stirred for 4–5 h and left overnight. The completeness of the reaction is controlled by 19F NMR data (signal 2 (–70 ppm) should be observed and signal of HFA (–83 ppm) should be absent.
The reaction mixture is washed with 6-8 l of 1 M HCl, the organic layer is separated, washed two times with equal volume of 5% HCl, 0.5 volume of conc. H2SO4 is added at 15÷20оС, the mixture obtained is heated up to 60÷70оС to distill off Me3SiF and then distilled under reduced pressure collecting the fraction boiling at 50÷70оС/20 torr. The distillate is redistilled to afford 1200 g (70%) of carbinol 2, b.p. 30оС/10 torr (lit. 136÷137°С [28–29]). 1H NMR δ: 3.6 (s, OH); 19F NMR δ: –70.5 (s, CF3).
2-(Dichloromethyl)-1,1,1,3,3,3-hexafluoropropan-2-ol (3).
Zn dust (36 g, 0.55 g-at) is added portionwise (6 g at ~1–5 minutes interval) at 20÷25оС to a mixture of 151 g (0.53 mol) 2 and a solution of 140 g (2.6 mol) NH4Cl in 500 ml of water under vigorous stirring, the reaction mixture is stirred for 1–1.5 h, acidified, heated up to distinct separation of phases, the under layer is separated and distilled over equal volume of conc. H2SO4. Further rectification of distillate yields 106 g (80%) of carbinol 3, b.p. 114÷115оС (lit. 115оС [29]). 1H NMR δ: 3.5 (br.s, 1H, OH), 5.7 (s, 1H, CHCl2); 19F NMR δ: –73.7 (s, CF3).
1,1,1-Trichloro-3,3,3-trifluoro-2-(trifluoromethyl)propyl acetate (4a).
Triethylamine (53.1 g, 0.525 mol) is added under stirring at 5÷10оС to a solution of 142.7 g (0.5 mol) carbinol 2 in 500 ml of MeCN, then 41.2 g (0.525 mol) of acetyl chloride are added dropwise, the mixture is stirred 3 h at 20÷25оС, poured into double volume of cold water, the under layer is separated, washed two times with double volume of cold water and distilled under reduced pressure (10–15 torr) over 10 g of P2O5, collecting the fraction that boils at 55÷65оС. Redistillation affords 142 g (87%) of ester 4a, b.p. 62÷63оС/10 torr. 1H NMR δ: 2.2 (s, CH3); 19F NMR δ: –65.0 (s, CF3).
1,1,1-Trichloro-3,3,3-trifluoro-2-(trifluoromethyl)propyl trifluoroacetate (4b).
Triethylamine (77.9 g, 0.8 mol) is added dropwise under stirring at 5÷10оС to a mixture of 200 g (0.7 mol) carbinol 2 and 220.5 g (1.05 mol) (CF3CO)2O, stirred for 2–3 h at 20÷25оС, volatile components of the reaction mixture are distilled off under reduced pressure (10–15 torr) into a trap (–78°C), the residue is distilled, collecting the fraction that boils at 50÷60оС. Redistillation affords 227 g (85%) of ester 4b, b.p. 60оС/10 torr. 19F NMR δ: –77.0 (s, 3F, CF3CO2), –65.8 (s, 6F, (CF3)2C).
1,1,1-Trichloro-3,3,3-trifluoro-2-(trifluoromethyl)propyl methane sulfonate(4c).
Triethylamine (167 g, 1.65 mol) is added under stirring at 5÷10оС to a solution of 427.5 g (1.498 mol) carbinol 2 in 1 l of MeCN, then 189 g (1.65 mol) of methanesulfonyl chloride are added dropwise, the mixture is stirred for 3–4 h at 20÷25оС, left overnight, poured into double volume of water, the under layer is separated, washed two times with double volume of water (if necessary small amount of acetone is added to prevent curdling) and distilled over 10 g of P2O5 collecting fraction boiling at 60÷80°C/0.1 torr. Redistillation there yields 508.3 g (93%) of sulfonate 4c, containing 4% 2, b.p. 80оС/0.1 torr. 1H NMR δ: 3.35 (s, CH3); 19F NMR δ: –64.5 (s, CF3).
1,1-Dichlorohexafluoroisobutylene (1).
Methane sulfonate 4c (300 g, 0.825 mol) is added dropwise under stirring at 150÷160оС to a suspension of 108 g (1.65 g-at) Zn dust activated by 8 ml of Me3SiCl in 300 ml of sulfolane accompanied by simultaneous distillation of olefin 1 containing 2–8% of 1-chlorohexafluoroisobutylene 5 and trace amounts of HMDS. The distillation of product obtained over conc. H2SO4 yields 153.8 g (80%) of olefin 1, b.p. 74оС (lit. 71÷74оС [30]). 19F NMR δ: –61.5 (s, CF3) (the product can be rectified if necessary).
1-Chlorohexafluoroisobutylene (5): 1H NMR δ: 5.4 (br.s, H); 19F NMR δ: –66.4 (m, 3F, CF3), –63 (m, 3F, CF3). Mass-spectrum (M/Z, reference): 198[M]+; 179[M–F]+; 163[M–Cl]+; 147[M–HCF2]+; 131[M–Cl–HCF]+; 129[M–CF3]+; 69[CF3]+(100%); 67[HCFCl]+; 48[CHCl]+.
Synthesis of compounds 7a-c, general procedure.
Dichlorohexafluoroisobutylene 1 (42 g, 0.18 mol) is added under stirring at 15÷20оС to a suspension of 100 g (0.658 mol) dry CsF in 150 ml of DMF. The reaction mixture is stirred for 2 h sustaining the temperature <30°C by external cooling. Then 0.2 mol of allyl bromide or bromoethyl acetate or 0.1 mol of sulfur monochloride (S2Cl2) is added under stirring at 20÷25оС. The reaction mixture is stirred for 2–3 h, left overnight, diluted with 350 ml of 5% HCl, the product is extracted with CH2Cl2, the extract is washed several times with 5% HCl, the solvent is evaporated and the residue is distilled. The purification of the product is achieved by rectification.
(CF3)3CCH2CH=CH2 (7a).
Obtained 32.8 g (70%), b.p. 76÷77оС (lit. 75÷76оС [31]). 19F NMR δ: –67 (s, CF3).
(CF3)3CCH2CH2OAc (7b).
Obtained 38.5 g (70%), b.p.141÷142оС (lit. 140÷142оС [32]). 19F NMR δ: –69 (s, CF3).
(CF3)3CSSSC(CF3)3 (7c).
Obtained 28.8 g (60%), b.p. 160оС (lit. 96÷100оС/50 torr [33-34]). 19F NMR δ: –65 (s, (CF3)3C).
(CF3)3CBr (7d).
Isobutylene 1 (42 g, 0.18 mol) is added under stirring at 15÷20оС to a suspension of 100 g (0.658 mol) dry CsF in 150 ml of MeCN. The reaction mixture is stirred for 4–5 h sustaining the temperature <30°C by external cooling. Then Br2 (28.8 g, 0.18 mol) is added at 5÷10оС. The mixture is stirred for 15 min and heated up to 70оС distilling the product into cooled receiver (0оС). The distillate is left overnight in refrigerator (~–20°C), the crystalline product is separated, mixed with equal volume of cold conc. H2SO4 and sublimed. Resublimation affords 32.3 g (60%) of bromide 7d, purity 90%. Sublimating temperature 35÷40оС (lit. m.p. 58÷59оС [35]). 19F NMR δ: –68 (s, (CF3)3C).
(E)-1-Bromo-6,6,6-trifluoro-5,5-bis(trifluoromethyl)hex-2-ene (7e).
Olefin 1 (84 g, 0.36 mol) is added under stirring at 15÷20оС to a suspension of 200 g (1.32 mol) dry CsF in 400 ml of DMF. The reaction mixture is stirred for 2 h sustaining the temperature <30°C by external cooling. Then, 92 g (0.43 mol) of 1,4-dibromobut-2-ene are added at 20÷25°C, the reaction mixture is stirred for 2–3 h, left overnight, diluted with 700 ml of 5% HCl, the product is extracted with CH2Cl2, the extract is washed several times with 5% HCl, the solvent is evaporated, the residue is distilled, collecting the fraction boiling at 40÷60оС/10 torr. The product is redistilled, collecting the fraction boiling at 50÷55°C/10 torr. The distillate obtained is left overnight at ~5оС, the crystalline precipitate of the initial 1,4-dibromobut-2-ene is filtered off to give 106 g of hexene 7e that is used in the next stage without further purification.
(E)-6,6,6-Trifluoro-5,5-bis(trifluoromethyl)hex-1,3-diene (8).
The mixture of 100 g (0.28 mol) 7e and 31.9 g (0.57 mol) of KOH in 150 ml of sulfolane is heated under stirring until the onset of vigorous an exothermal reaction that leads to intensive boiling of mixture. After spontaneous boiling is over the mixture is stirred and heated under reflux for 30 min, the products volatile at 80÷100°C are distilled, the organic layer of distillate is separated and distilled over KOH. Further rectification affords 45.7 g (60%) of diene 8, b.p. 97оС. Found (%): C, 35.38; H, 1.84; F, 63.08. C8H5F9. Calculated (%): C, 35.31; H, 1.85; F, 62.84. 19F NMR δ: –68 (s, (CF3)3C).
The reaction of olefin 1 with MeOH in the presence of bases; the preparation of ether 9 and ester 19.
Olefin 1 (10 g, 43 mmol) is added gradually under stirring to a solution of 2.83 g KOH in 12 ml МеОН, on the morrow of an exothermal reaction the mixture was stirred for 0.5 h, washed with aq. HCl, organic layer is separated, dried over MgSO4 and filtered to give 8 g of a mixture containing according to chromatography-mass spectrometry data 84% of ether 9 (19F NMR spectrum is identical to that described in [36]), yield 76% taking into account the coversion of olefin 1, 12% of olefin 1 and 4% of ester 19. Essentially the same composition of the reaction products is obtained in the reaction of 1 with MeOH in the presence of KF (3 eq.).
Mass-spectrum 9 (M/Z, reference): 228[M]+; 209[M–F]+; 193[M–Cl]+; 178[C4F6O]+; 159[C4F5O]+; 150[C3F6]+; 129[C3HF4O]+; 109[C3F3O]+; 91[C3HFCl]+; 81[C2F3]+; 71[C3FO]+; 69[CF3]+; 63[CClO]+.
Under the action H2SO4 the ether 9 is quantitatively converted into methyl α-hydrohexafluoroisobutyrate 19 (according to 19F NMR and mass-spectrometry data).
2,2-Dichloro-3,3-bis(trifluoromethyl)oxirane (10).
KOH (14.56 g, 0.26 mol) is added to 50 g (0.175 mol) of carbinol 2, the mixture is stirred for 0.5–1 h, gradually heated until the onset of an exothermal reaction that leads to intensive boiling of mixture. The boiling is kept under reflux for 30 min and the reaction product is distilled. The organic layer of distillate is separated and distilled over P2O5 to give 35 g (87%) of oxirane 10, b.p. 69÷70оС (lit. 68÷70оС [12]). 19F NMR δ: –69.4 (s, CF3).
2-Chloro-3,3-bis(trifluoromethyl)oxirane (11).
KOH (23 g, 0.41 mol) is added to 69 g (0.275 mol) of carbinol 3, within several minutes exothermal reaction begins. The temperature of mixture is controlled by external cooling to prevent its boiling. When the exothermal reaction ceases the mixture is boiled under reflux for 30 min, the reaction product is distilled. The distillate is redistilled over P2O5 to give 48 g (81%) of oxirane 11, b.p. 52÷53оС. 1H NMR δ: 5.4 (br.s, H); 19F NMR δ: –75.2 (m, 3F, CF3), –68.6 (m, 3F, CF3).
2,3,3,3-Tetrafluoro-2-(trifluotomethyl)propanoyl fluoride (13).
The mixture of 16.27 g (0.28 mol) dry KF, 20 g (0.07 mol) of carbinol 2 and 50 ml of dehydrated diglyme is gradually heated under stirring with reflux condenser before the beginning of exothermic reaction. The gaseous reaction products are condensed in a trap (–78°C). The condensate is distilled to give 7.5 g (50%) of acyl fluoride 13, b.p. 2÷4оС (lit. 2÷3оС [37]). 19F NMR δ: –183.4 (m, 1F, CF(CF3)2), –77.2 (d, 6F, 3JFF = 5.6 Hz, CF(CF3)2), 29.6 (m, 1F, COF).
2-Chloro-3,3,3-trifluoro-2-(trifluotomethyl)propanoyl chloride (14).
Oxirane 10 (67 g, 0.27 mol) is added under stirring to a suspension of 0.76 g (0.013 mol) dry KF in 30 ml of sulfolane, the mixture is heated under reflux for 30 min, the reaction product is distilled using Vigreux column. The degree of isomerization 10 into 14 is controlled by 19F NMR: 10 (δ: –67.5); 14 (δ: –70.1). There is obtained 60.5 g (90%) of acyl chloride 14, b.p. 68÷69оС (lit. 70оС [5]). 19F NMR δ: –70 (s, CF3).
2-Chloro-3,3,3-trifluoro-2-(trifluotomethyl)propanoyl fluoride (15).
The mixture of 60 g (0.241 mol) acyl chloride 14, 35 g (0.6 mol) of dry KF and 30 ml of sulfolane is stirred and heated under reflux with Heckel column and nozzle of full condensation gradually collecting the fraction boiling at 35÷50оС. The degree of conversion 14 into 15 is controlled by 19F NMR: 14 (δ: –70.2); 15 (δ: –71.5). The distillation of product obtained yields 42 g (75%) of 15, b.p. 34÷35оС (lit. 34оС [5]). 19F NMR δ: –71.7 (d, 6F, 4JFF = 11 Hz, CF3), 32.4 (m, 1F, COF).
Methyl 2,3,3,3-tetrafluoro-2-(trifluoromethyl)propanoate (16).
Oxirane 10 (40 g, 0.16 mol) is added under stirring to a suspension of 28 g (0.48 mol) dry KF in 100 ml of MeOH. Upon completion of exothermal reaction the mixture is heated under reflux for 1 h, cooled, poured into cold water, the under layer is separated, washed with cold water and distilled over P2O5. Further rectification gives 29 g (80%) of ester 16, b.p. 75÷76оС (lit. 76÷77оС [38]). 1H NMR: δ 4.3 (br.s, OCH3); 19F NMR:δ –183 (m, 1F, CF(CF3)2), –76.5 (d, 6F, CF(CF3)2).
The reaction of 2,2-dichloro-3,3-bis(trifluoromethyl)oxirane 10 with KBr.
Oxirane 10 (10 g, 40 mmol) is gradually added to a suspension of 14.3 g (120 mmol) KBr in 50 ml of diglyme, Upon completion of exothermic reaction the mixture is stirred at 75÷83оС/1.5 h. According to 19F NMR data the reaction mass contains a mixture of α-bromo-, α-chloro- and α-hydrohexafluoroisobutyryl halogenides in molar ratio 4:2:1 correspondingly. Methanol (8 ml) is added to the reaction mass, the mixture is stirred for 10 min, washed with diluted HCl–acid, the organic layer is separated and dried over MgSO4 to give 9.95 g of a mixture containing 90% of esters (CF3)2CBrCO2Me (17), (CF3)2CClCO2Me (18) and (CF3)2CHCO2Me (19) in molar ratio 1:1:0.8.
Mass-spectrum 17 (M/Z, reference): 288[M]+; 257[C4BrF6O]+; 229[C3BrF6]+; 210[C3BrF5]+; 191[C3BrF4]+; 179[C2BrF4]+; 178[C4F6O]+; 169[C3BrF2O]+; 160[C2BrF3]+; 159[C4H3F4O2]+; 150[C3F6]+; 131[C3F5]+(100%); 129[CBrF2]+; 112[C3F4]+; 100[C2F4]+; 93[C3F3]+; 81[C2F3]+; 71[C3FO]+, 69[CF3]+; 59[C2H3O2]+; 47[CFO].
Mass-spectrum 18 (M/Z, reference): 213[C4ClF6O]+; 185[C3ClF6]+; 166[C3ClF5]+; 159[C4H3F4O2]+; 150[C3F6]+; 131[C3F5]+; 116[C2ClF3]+; 100[C2F4]+; 85[CClF2]+; 81[C2F3]+; 69[CF3]+(100%); 59[C2H3O2]+; 47[CFO].
Mass-spectrum 19 (M/Z, reference): 225[M+Me]+; 211[M+H]+(100%); 191[M–F]+; 179[C4HF6O]+; 170[C5H2F4O2]+; 159[C4H3F4O2]+; 150[C3F6]+; 140[C4H3F3O2]+; 113[C3HF4]+; 91[C3HF2O]+; 81[C2F3]+; 69[CF3]+; 59[C2H3O2]+.
The reaction of oxirane 10 with 0.5 eq. SbF5.
2-Chloro-2-fluoro-3,3-bis(trifluoromethyl)oxirane (21).
A) Antimony pentafluoride (13 g, 0.06 mol) is added portionwise under stirring at 20÷25оС to 30 g (0.12 mol) of oxirane 10, the mixture is stirred for 1h, poured onto crashed ice, acidified with HCl–acid, the under layer is separated, washed with diluted HCl and distilled over conc. H2SO4 to give 19 g of distillate that contains 25% of oxirane 21 and 63% of carbinol 12 (according to GLC and 19F NMR-spectrometry data).
B) The reaction mixture prepared as described above is heated slowly with simulteneous distillation of the reaction product into reciever (–78°). Rectification of distillate affords 22 g (80%) of oxirane 21, b.p. 34.5÷35.5оС. Found (%): С, 20.53; Cl, 14.68; F, 56.73. C4СlF7O. Calculated (%): С, 20.67; Cl, 15.25; F, 57.20. 19F NMR δ: –88 (m, 1F, CFCl), –70 (s, 6F, CF3). Mass-spectrum (M/Z, reference): 216[M–O]+, 197[M–Cl]+, 181[C4F7]+, 169[C3F7]+, 147[C3ClF4]+, 119[C2F5]+, 93[C3F3]+, 85[CClF2]+, 69[CF3]+(100%), 47[CFO]+.
Mass-spectrum of carbinol 12 (M/Z, reference): 252[M]+, 233[M–F]+, 217[M–Cl]+, 197[C4F7O]+, 185[C3F6Cl]+, 163[C3ClF4O]+, 147[C3F5O]+, 119[C2F5]+, 85[CClF2]+, 69[CF3]+(100%), 51[CHF2]+, 50[CF2]+.
The reaction of oxirane 10 with 1 eq. SbF5.
A) Antimony pentafluoride (26 g, 0.12 mol) is added portionwise under stirring at 20÷25оС to 30 g (0.12 mol) of oxirane 10. The viscous mixture is stirred for 1 h, poured onto crashed ice, HCl–acid is added to dissolve antimony oxide, the under layer is separated, washed with diluted HCl–acid and distilled over conc. H2SO4 to give 26 g (85%) of carbinol 12, b.p. 74÷75оС (lit. 73÷74оС [29]). 1H NMR δ: 3.7 (br.s, OH); 19F NMR δ: –74.3 (t, 6F, 4JFF = 7.5 Hz, CF3), –62.2 (sept., 2F, CF2Cl).
B) The reaction mixture odtained as described above is slowly heated up to 180÷190оС accompanied with simulteneous distillation of the reaction ptoducts into cooled reciever (–78°C). The distillate obtained (19 g) contains 36% of oxirane 21, 33% of perfluoroisobutylene oxide (22), 29% of acyl fluoride 13 and negligible amounts of other reaction products such as carbinol 12 (2%) and trace amounts of chlorine.
References
- Yu.V. Zeifman, E.G. Ter-Gabrielyan, N.P. Gambaryn, I.L. Knunyants Russ.Chem.Rev., 1984, 53, 256-273.
- N.P. Gambaryan, E.M. Rokhlin Russ.Chem.Rev., 1986, 55, 480-494.
- I.L. Knunyants, V.R. Polishchuk Russ.Chem.Rev., 1976, 45, 574-592.
- B.L. Dyatkin, N.I. Delyagina, S.R. Sterlin Russ.Chem.Rev., 1976, 45, 607-614.
- I.L. Knunyants, Yu.A. Cheburkov, M.D. Bargamova Bull.Acad.Sci.USSR, Div.Chem.Sci., 1963, 12, 1265-1268.
- D.C. England, C.G. Krespan J.Am.Chem.Soc., 1965, 87, 4019-4020.
- M.S. Raasch J.Org.Chem., 1970, 35, 3470-3483.
- E.M. Rokhlin, E.G. Abduganiev, U. Utebaev Russ.Chem.Rev., 1976, 45, 593-606.
- S. Ebnesajjad Fluoroplastics, Vol. 1, 2nd Ed., pp. 48-75, 2015.
- C.M. Timperley Fluorine Chemistry at the Millennium Fascinated by Fluorine, Ed. R.E. Banks, pp. 499-538, 2000.
- C.M. Timperley J.Fluor.Chem., 1999, 94, 37-41.
- Yu.V. Zeifman Bull.Russ.Acad.Sci., Div.Chem.Sci., 1992, 41, 370-373.
- A.A. Tyutyunov, A.V. Sin’ko, S.M. Igumnov, O.A. Mel’nik, Ya.S. Vygodskii, E.V. Khaidukov, V.I. Sokolov Dokl.Chem., 2016, 467, 88-90.
- R.E.A. Dear, E.E. Gilbert, J.J. Murray Tetrahedron, 1971, 27, 3345-3355.
- H.B. Dykstra, J.F. Lewis, C.E. Boord J.Am.Chem.Soc., 1930, 52, 3396-3404.
- J.W. Cornforth, R.H. Cornforth, K.K. Mathew J.Chem.Soc., 1959, 112-127.
- R.A. Bekker, G.V. Asratyan, B.L. Dyatkin, I.L. Knunyants Dokl.Akad.Nauk SSSR, 1972, 204, 606-609.
- R.A. Bekker, G.V. Asratyan, B.L. Dyatkin Zhur.Org.Khim., 1973, 9, 1635-1640.
- A.N. Nesmeyanov, K.A. Pecherskaya, G.Ya. Uretskaya Izv.Acad.Nauk USSR, Otdel.Khim.Nauk, 1948, 240-245.
- J.H. Clark Chem.Rev., 1980, 80, 429-454.
- W. Reeve Synthesis, 1971, 131-138.
- T.S. Snowden ARKIVOC, 2012, 2, 24-40.
- Yu.A. Cheburkov, M.D. Bargamova Bull.Acad.Sci.USSR, Div.Chem.Sci., 1967, 16, 801-806.
- I.L. Knunyants, V.V. Shokina, V.V. Tyuleneva, T.N. Razumeeva Bull.Acad.Sci.USSR, Div.Chem.Sci., 1972, 21, 1085-1088.
- J.T. Hill J.Fluor.Chem., 1977, 9, 97-112.
- D.E. Morin US Pat. № 3,213,134 (1965).
- F.J. Pavlik US Pat. № 3,385,904 (1968).
- R. Filler, R.M. Schure J.Org.Chem., 1967, 32, 1217-1219.
- R.E.A. Dear Synthesis, 1970, 361-362.
- V.V. Tyuleneva, L.A. Rozov, Yu.V. Zeifman, I.L. Knunyants Bull.Acad.Sci.USSR, Div.Chem.Sci., 1975, 24, 1042-1045.
- N.I. Delyagina, E.Ya. Pervova, I.L. Knunyants Bull.Acad.Sci.USSR, Div.Chem.Sci., 1972, 21, 326-329.
- I.L. Knunyants, N.I. Delyagina, B.L. Dyatkin, I.Ya. Aliev USSR certificate of authorship № 379556 (1973).
- C.G. Krespan, D.C. England J.Org.Chem., 1968, 33, 1850-1854.
- D.C. England US Pat. № 3,544,591 (1970).
- B.L. Dyatkin, A.A. Gevorkyan, I.L. Knunyants Bull.Acad.Sci.USSR, Div.Chem.Sci., 1965, 14, 1833-1835.
- D.I. Rossman, A.J. Muller J.Fluor.Chem., 1993, 60, 61-68.
- I.L. Knunyants, V.V. Shokina, V.V. Tyuleneva, Yu.A. Cheburkov, Yu.E. Aronov Bull.Acad.Sci.USSR, Div.Chem.Sci., 1966, 15, 1764-1766.
- R.D. Smith, F.S. Fawcett, D.D. Coffman J.Am.Chem.Soc., 1962, 84, 4285-4288.
Recommended for publication by Prof. V. Kornilov
Fluorine Notes, 2018, 121, 9-10
