Received: October 2019
DOI 10.17677/fn20714807.2019.05.05
Fluorine Notes, 2019, 126, 9-10
SYNTHESIS OF DIFLUOROMETHYL KETONES
Irina V. Sandulenko
A.N. Nesmeyanov Institute of Organoelement Compounds, Russian Academy of Sciences, Vavilov St. 28, Moscow, 119991, Russian Federation
e-mail: ira17.rock@mail.ru
Abstract: The minireview focused on the basic methods for the synthesis of difluoromethylketones, which are of interest for medicinal chemistry and a valuable building blocks for other substituted α,α-difluoroketones preparation. Some approaches to a difluoroacetyl moiety introduction into chemical compounds of various classes are presented.
Keywords: difluoromethyl ketones, difluoromethylation, difluoroacylation.
Introduction
Over a past few decades synthetic organofluorine chemistry involves more in drug design due to the benefits that fluorine atom brings in pharmacological profile of a biologically active molecule [1]. In particular, α-gem-difluorinated carbonyl compounds are of great interest for medicinal chemistry [2; 3]. Whilst trifluoromethyl ketones are synthetically available in most cases [4], the synthesis of α,α-difluoroketones is challenge up to now [5]. Our review provides the basic methods for difluoromethyl ketones synthesis. These structures with a difluoroacetyl moiety due to functionalization of the C-H bond of CF2H [6,7] via cross-coupling reactions [8-10] particularly proved to be a valuable synthetic intermediates on the way to other substituted α,α-difluoroketones.
The review is structured, each section covers the synthetic approach and specifically describes how the difluoromethyl fragment was introduced into substrate: with or without the carbonyl carbon atom, by electrophilic fluorination at the α-position or defluorination of the CF3 fragment.
1. Difluoroacylation
In early attempts to obtain difluoromethylketones the organometallic substrates and some difluoroacetic acid derivatives as difluoroacylation agents were actively used. For instance, a series of acetylenic difluoromethyl ketones 2 as objects for studying the regio- and stereoselectivity of cuprate reagents addition was synthesized in good yields starting from lithium acetylides 1 and ethyl difluoroacetate in the presence of boron trifluoride etherate [11].

|
Lithium acetylide 1, R |
Yield of 2, % |
|
Ph |
77 |
|
n-C4H9 |
74 |
|
n-C7H15 |
71 |
|
n-C8H17 |
66 |
|
n-C10H21 |
64 |
Scheme 1.
The authors [2] account for relatively poor yields of difluoromethylketones 5 obtained with ethyl difluoroacetate from aromatic organolithium compounds 3 as a result of side reactions with the initially formed adduct 4.
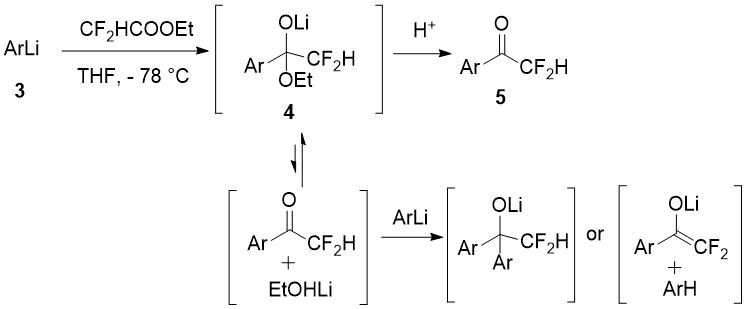
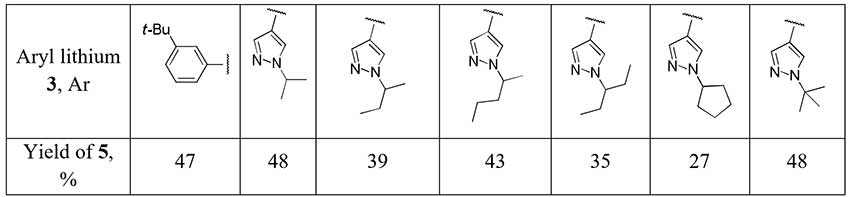
Scheme 2.
In the late 80s Japanese scientists described a synthesis of difluoromethylketones 8 in moderate to good yields via 2,2-difluoroalkenylboranes 7 oxidation [12].
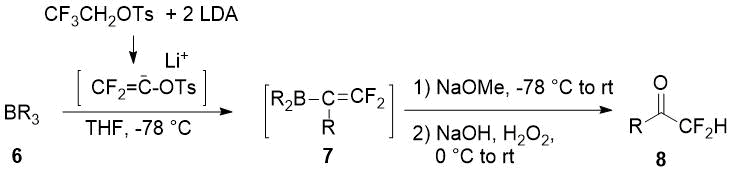
|
Trialkylborane 6, R |
Yield of 8, % |
|
-(CH2)4Ph |
81 |
|
-CH2CH(CH3)Ph |
77 |
|
10-Pinanyl |
63 |
|
Cyclooctyl |
63 |
|
Bicyclo[2.2.1]hept-1-yl |
67 (endo/exo diastereomers 92/8) |
|
-C(Prn)=CH-Prn |
27 |
Scheme 3.
Aryl difluoromethyl ketones 10 can be obtained in good yields using difluoroacetonitrile in superacid-promoted Houben-Hoesch reaction with some activated arenes 9 [13].
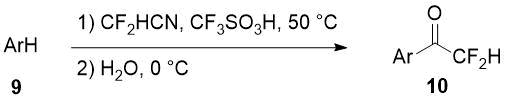
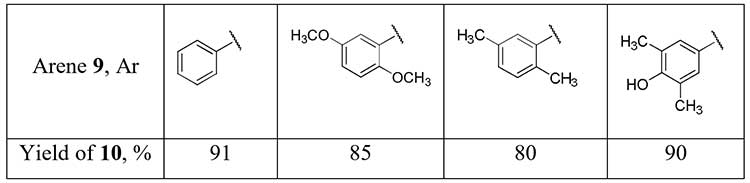
Scheme 4.
1,1,2,2-Tetrafluoroethyl-N,N-dimethylamine (11, TFEDMA) activated with BF3·Et2O proved to be a difluoroacetyl transfer agent for arenes and heteroarenes [14]. The authors succeded to obtain some difluoroacetyl derivatives of electron-enriched heterocycles (12) using this reagent.
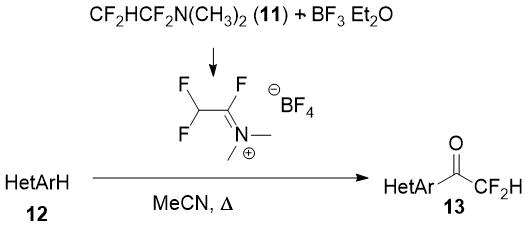
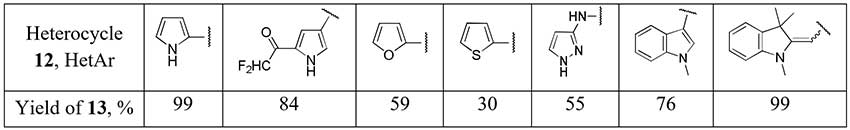
Scheme 5.
Both good regioselectivity and yields of the target difluoromethylketones 15 obtained from arenes with electron-donating substituents (14) were achieved using microwave irradiation. It was also able to prepare the difluoroacylated arenes 15 in good yields in reaction of aryllithiums with N,N-dimethyldifluoroacetamide [14] (hydrolyzed TFEDMA, Scheme 6, method B).
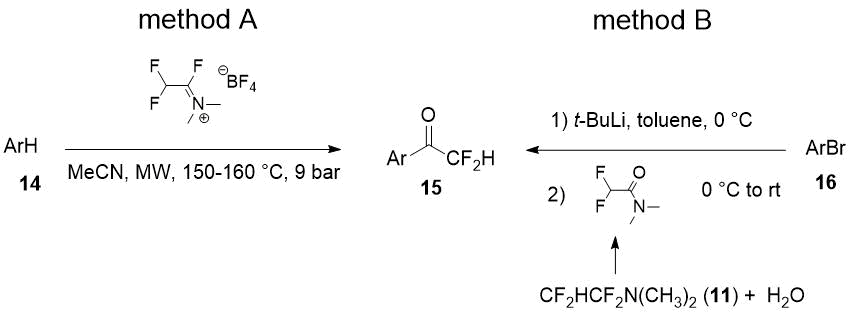
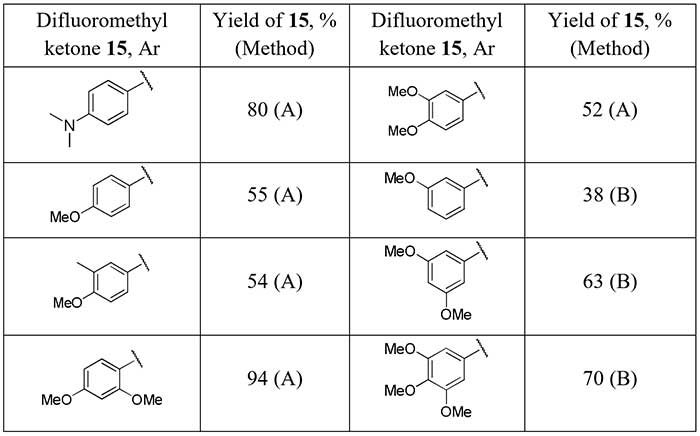
Scheme 6.
It was found previously that TFEDMA also difluoroacetylates some cyclic 1,3-diketones with formation of corresponding 2-difluoroacetyl derivatives via the intermediate adduct 17 [15].
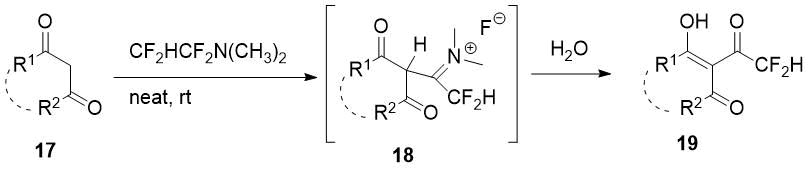
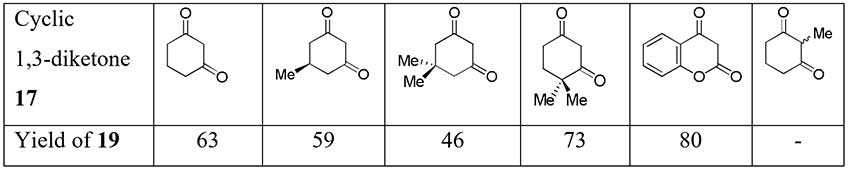
Scheme 7.
A series of difluoroacetophenones 22 has been synthesized by cleavage of α,α-difluoro-β-keto esters 21 prepared by coupling of readily available aryl boronates 20 with bromodifluoroacetamide and carbon monoxide [16]. The coupling process is catalyzed by [Pd(PPh3)4] with Xantphos as a secondary ligand and CuI as a cocatalyst. Decarboxylation of ethyl esters 21 was carried out without intermediate purification and the authors supposed the one-pot synthesis of difluoroacetophenones by their method is possible.
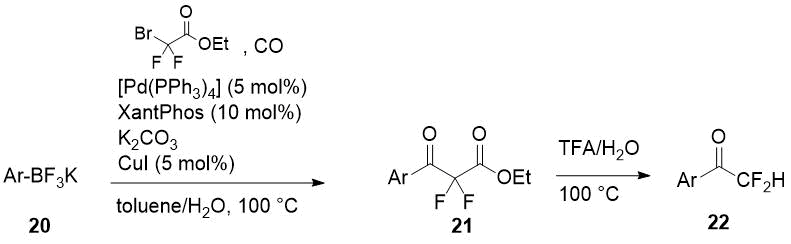
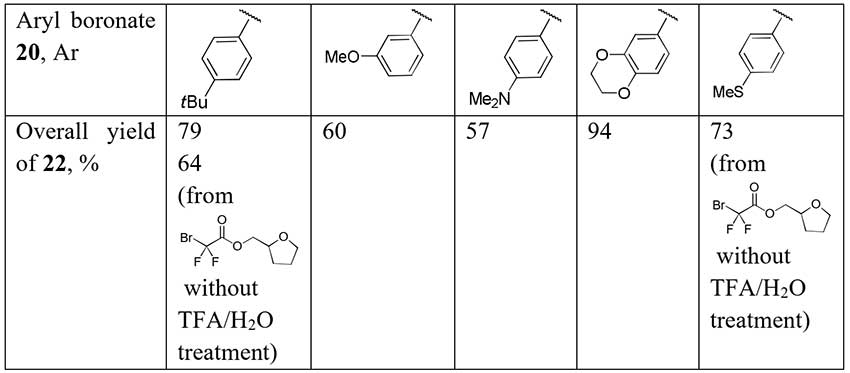
Scheme 8.
2. Defluorination
Difluoromethyl ketones can be prepared by hydrolysis of corresponding 2,2-difluoro enol silyl ethers. Several approaches have been developed for the synthesis of the last ones. The synthesis of 25 is the most interesting example of difluoromethyl ketone preparation from these intermediates. Ketone 25 was obtained in overall yield of 43% from acylsilane 23a or in almost quantitative yield from 23b in result of successive transformations including trifluoromethylation with the Ruppert-Prakash reagent, the Brook rearrangement with fluoride elimination, and the final hydrolysis [17].
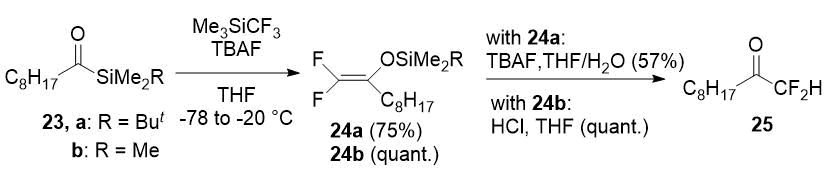
Scheme 9.
Selective Mg-promoted defluorination of trifluoromethyl ketones 26 with 2,2-difluoro
enol silyl ethers 27 formation was first carried out by Amii and coworkers [18],
and two years later Prakash used this synthetic equivalents of α,α-difluorinated enolates to produce
a small series of aromatic/heteroaromatic difluoromethyl ketones 28 [19]. Just one
example of an aliphatic difluoromethylketone synthesis by this method in moderate yield is given
in the report.
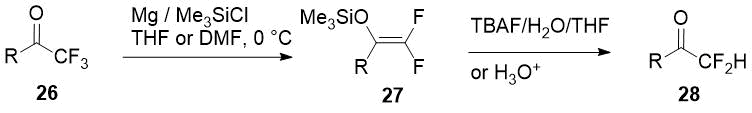
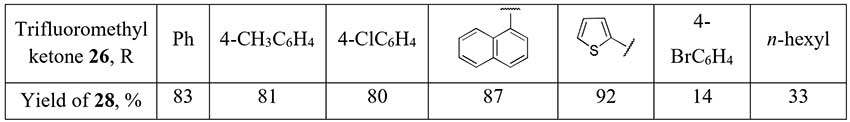
Scheme 10.
3. Difluoromethylation
Another method for the synthesis of difluoromethyl aryl ketones 28 via corresponding enol silyl ethers 29 with PhSCF2SiMe3 [difluoro(phenylsulfanyl)methyl]trimethylsilane] as source of the difluoromethyl moiety was developed by Pohmakotr et al. [20]. Silyl ethers 30 is formed in mixture with alcohols 31 and easily separated chromatographically. Subsequent oxidation of 30 with m-chloroperoxybenzoic acid (m-CPBA) and pyrolysis of sulfoxides 32 leads to 2,2-difluoro enol silyl ethers 33 and the target ketones 34 after hydrolysis in good overall yields.
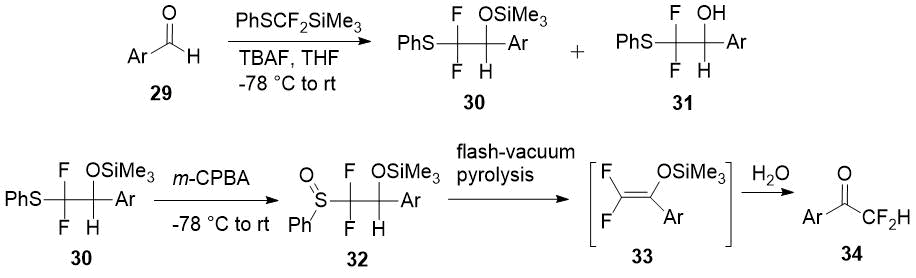
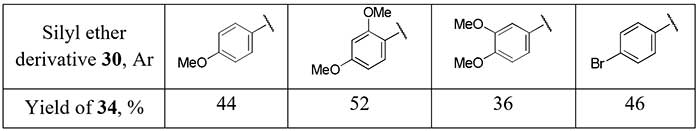
Scheme 11.
An another way to difluoromethyl ketones using the same reagent (PhSCF2SiMe3) consists in addition of the last one to Weinreb amides 35 [21] with subsequent reductive cleavage of the phenylsulfonyl group. A thorough optimization of the reaction conditions at both synthesis stages allowed to obtain a series of difluoromethylketones 37 with significant structural diversity.
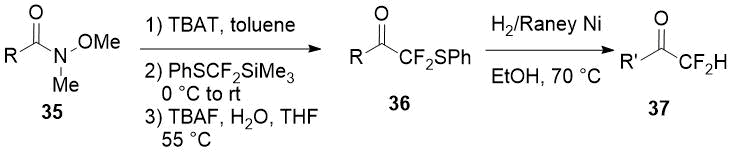
|
Weinreb amide 35, R |
Overall yield of 37 (R’ = R), % |
Weinreb amide 35, R |
Overall yield of 37, % |
|
3,4,5-(MeO)3C6H2 |
75 |
3-ClC6H4 |
82 (R’ = R) |
|
3,5-(MeO)2C6H3 |
84 |
β-naphthyl |
82 (R’ = R) |
|
3-MeOC6H4 |
81 |
n-C7H15 |
86 (R’ = R) |
|
3-MeC6H4 |
69 |
2-thienyl |
76 (R’ = R) |
|
4-tBuC6H4 |
83 |
C6H5CH=CH |
53 (R’ = C6H5CH2CH2) |
|
Ph |
84 |
4-ClC6H4CH=CH |
49 (R’ = 4-ClC6H4CH2CH2) |
Scheme 12.
Previously aromatic difluoromethylketone 40 was prepared in four steps from aldehyde 38 using PhSO2CF2H as a difluoromethylating agent by Hu et al. [22]. The key intermediate of the process is 2,2-difluorinated enol benzoate 39, and such benzoates, according to the authors, are quite stable in contrast to 2,2-difluorinated silyl enol ethers and more attractive difluoromethyl-containing building blocks.
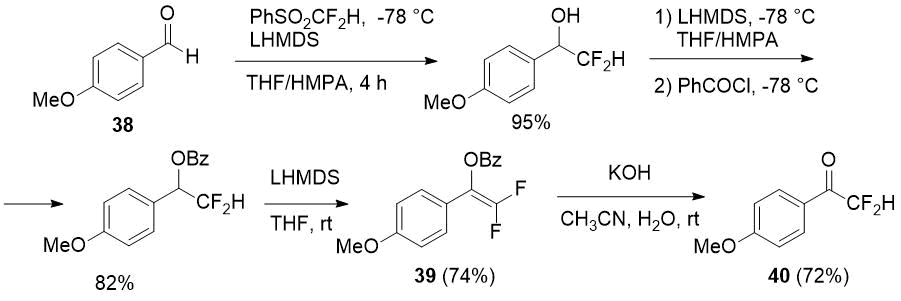
Scheme 13.
Chen et al. [23] showed a series of difluoromethylketones 43 could be obtained easily by heating adduct of aldehydes 41 with potassium phenylsulfonyl difluoroacetate (PhSO2CF2COOK) with heat-promoted removal of PhSO2 group from intermediate 42.
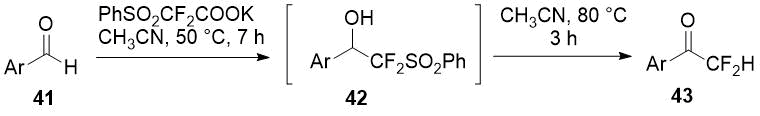
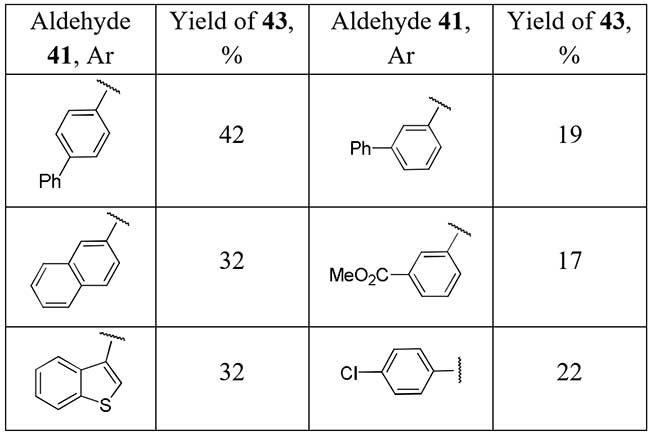
Scheme 14.
Piettre et al. [24] described a method for some aliphatic and aromatic difluoromethylketones (46) synthesis starting from aldehydes 44. A three-step approach uses lithium salt of diethyl difluoromethylphosphonate as a source of the difluoromethyl fragment and involves addition of the last one to aldehyde 44, oxidation of the obtained alcohols 45 with pyridinium dichromate (PDC) or DMSO/oxalyl chloride system (Swern oxidation) and final dephosphonylation.
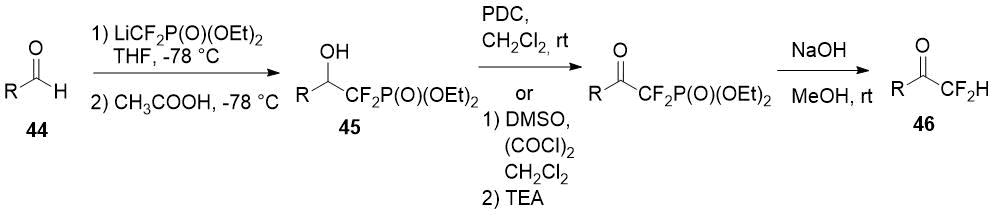
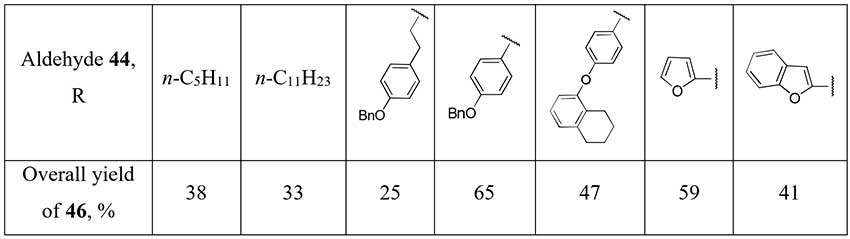
Scheme 15.
A rare example of synthesis of the structures with a difluoroacetyl function using a specific difluoromethylation
metal complex reagent is the work of Chinese scientists [25]. Having available N-heterocyclic carbene
ligated Ag-CF2H complex (48) Gu et al. succeed to obtain a significant
series of difluoromethyl ketones 49 in good yields, including aliphatic derivatives,
starting from acid chlorides 47.
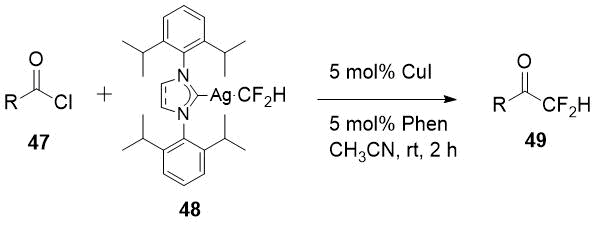
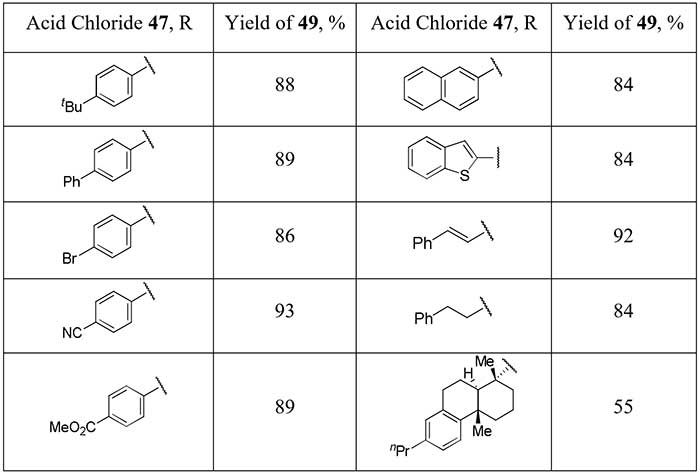
Scheme 16.
The one-pot method for the difluoromethylation of carboxylic acids 50 by addition in situ generated difluorinated phosphorus ylide to acid chlorides 51 seems to be rather promising as the method for difluoromethyl ketones 53 preparation [26]. However, either a substrate with a trisubstituted α-carbon atom or a nucleophilic additive (DMF) into reaction mixture is required for selective formation of monophosphonium salt 52.
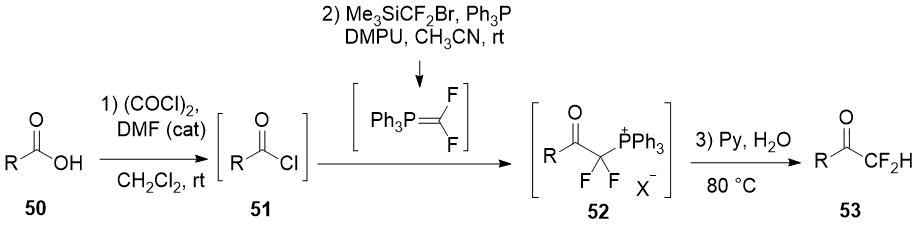
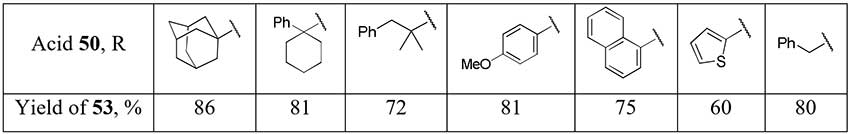
Scheme 17.
The same authors showed how difluoromethylketones can be prepared by one pot method starting from aldehydes and the same difluoromethylating reagent (Me3SiCF2Br) via easily generated in high yield iododifluoromethylated silyl ethers [27] giving the synthesis of ketone 54 as an example.
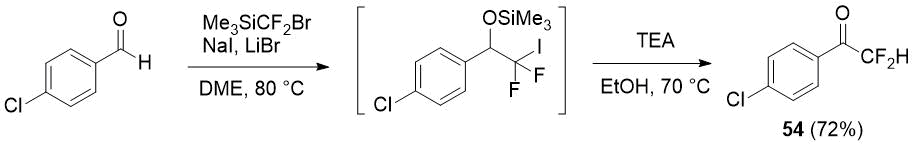
Scheme 18.
4. Electrophilic fluorination
In 1987 arylacetylenes were found to be easily fluorinated under mild conditions with cesium fluoroxysulphate [28], including phenylacetylene (55) to form difluoroacetophenone (56) in a mixture with dimethoxy derivative 57.
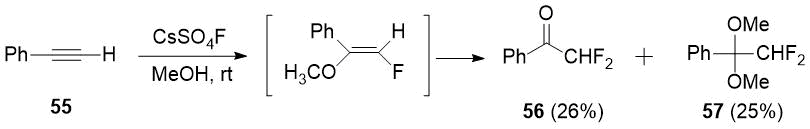
Scheme 19.
Later the same authors [29] described a method of synthesis of 56 as the only product. The reaction was carried out in the presence of water using Selectfluor® [1-chloromethyl-4-fluoro-1,4-diazoniabicyclo[2.2.2]octane bis(tetrafluoroborate)] as a fluorinating agent. A year later, the authors [30] managed to slightly increase the yield of difluoroacetophenone (56) when used Accufluor® [1-fluoro-4-hydroxy-1,4-diazoniabicyclo[2.2.2]octane) bis-(tetrafluoroborate)].
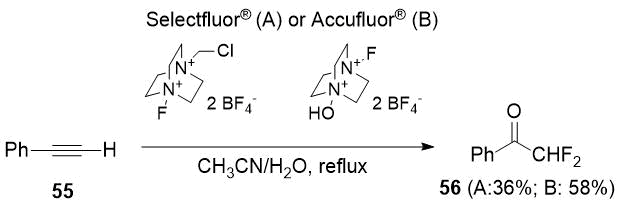
Scheme 20.
Imines 58 easily available from primary alkyl amines and ketones or aldehydes could be fluorinated with (CF3SO2)2NF (N-fluoro-bis[(trifluoromethyl)sulfonyl]imide) giving mixture of monofluoro 59 and difluoro products 60 after an acid hydrolysis [31]. Difluoromethyl ketones 60 can be obtained as a sole products using excess of the fluorinating agent.
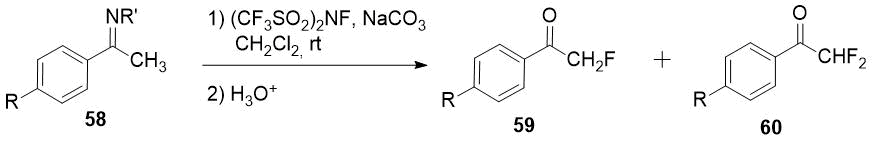
|
Imine 58 |
(CF3SO2)2NF, eq |
Yield of 59, % |
Yield of 60, % |
|
|
R |
R’ |
|||
|
H |
n-Pr |
2.4 0.67 |
0 33 |
82 15 |
|
4’-Me |
n-Pr |
2.4 0.67 |
0 36 |
83 16 |
|
4’-MeO |
n-Bu |
2.4 0.67 |
0 26 |
70 17 |
|
4’-Br |
n-Bu |
2.4 0.67 |
0 30 |
78 16 |
Scheme 21.
Selectfluor also allowed to get difluoromethylketones 62 in some cases as the only products
from enamines 61 [32].
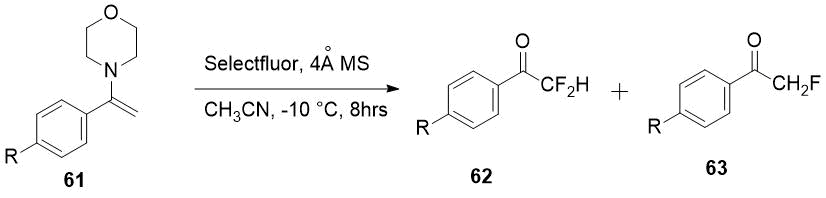
|
Enamine 61, R |
Products ratio 63/62 |
Yield of 62, % |
|
H |
1:11 |
74 |
|
MeO |
1:2.8 |
64 |
|
NO2 |
0:100 |
95 |
|
EtOOC |
0:100 |
72 |
Scheme 22.
The one-pot difluorination strategy of the α-monosubstituted carbonyl compound followed by removal of the blocking group preventing the overfluorination is applied in Pattison group [33] for selective difluoromethyl ketones preparation. Trifluoroacetyl group was used as a protection and as a promoter of the second stage of diketones 64 fluorination and it was easily removed. Optimization of conditions of 2,2-difluorinated 1,3-diketones 65 cleavage, avoiding self-aldol reaction, lead the authors to significant series of the target difluoromethyl ketones 66 in good yields.
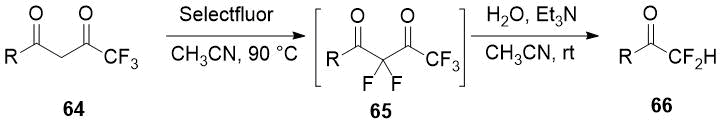
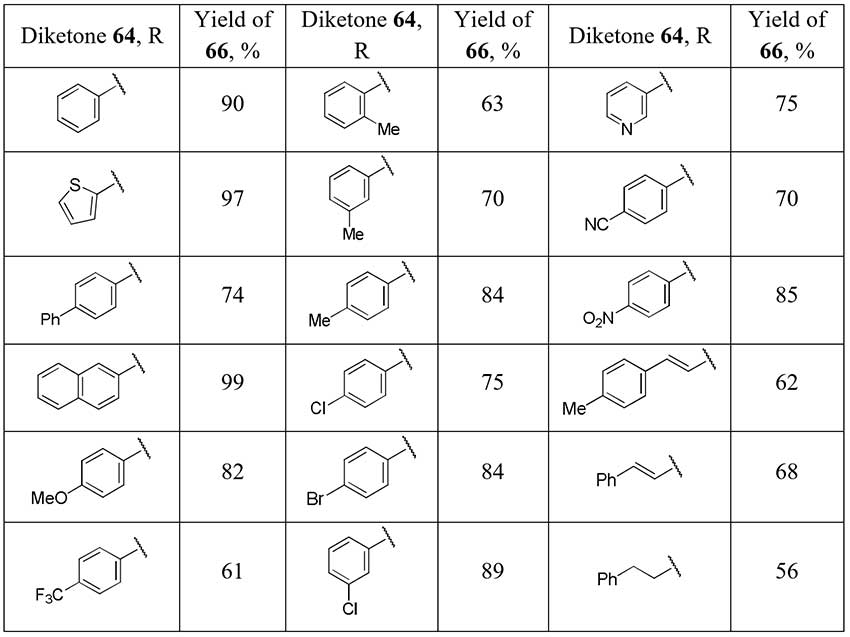
Scheme 23.
A similar strategy of difluorination followed by decarboxylation is applied by Li and co-workers to commercially available β-ketoacids 67 [34] for preparation of aryl and heteroaryl difluoromethyl ketones 68 in good to moderate yields.
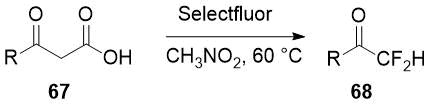
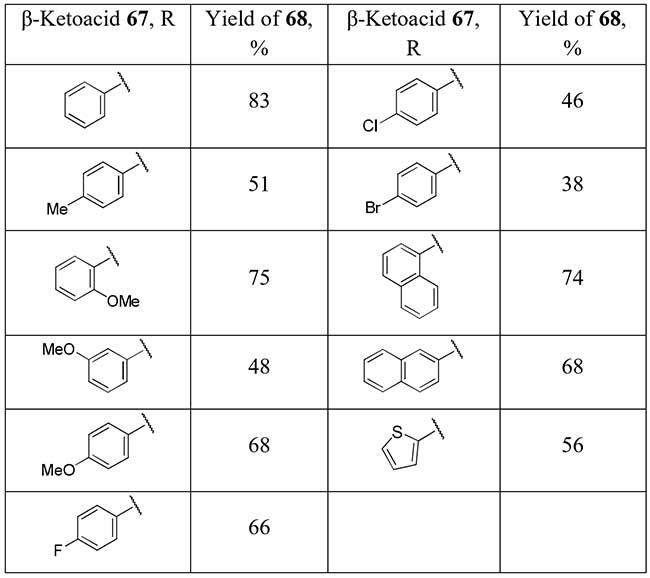
Scheme 24.
5. Miscellaneous
In an attempts to find an appropriate oxidizing agent for fluoroalkyl-substituted secondary alcohols, the authors [35] gave an example of the synthesis of difluoroacetophenone (56) in high yield using Oxone®/sodium 2-iodobenzenesulfonate additive as an oxidizing system.
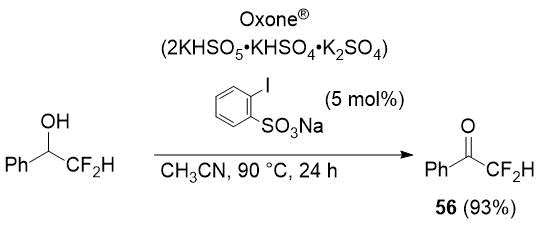
Scheme 25.
In a number of studies the same difluoroacetophenone (56) was obtained by removing the α-functional group from α-substituted α,α-difluoroacetophenones, for instance, by reduction of ready available 2,2-difluoro-2-iodo-1-phenylethanone (69) [36] or decarboxylation of 2,2-difluoro-3-oxo-3-phenylpropanoic acid (70) [37] which could also be prepared easily.
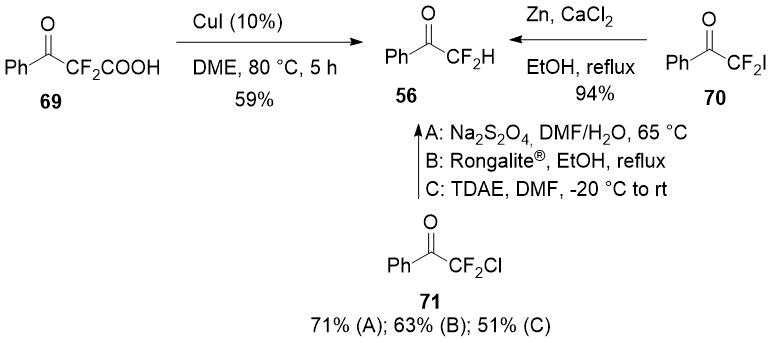
Scheme 26.
In developing a new strategies for the synthesis of difluoromethyl ketones by dehalogenation of halodifluoromethyl ketones with aromatic and heterocycle moieties French scientists [38] obtained difluoroacetophenone (56) from 2-chloro-2,2-difluoro-1-phenylethanone (71) with 51-71% yields depending on the organic reducing agent used (sodium hydroxymethanesulfinate (Rongalite®), sodium dithionite, tetrakis(dimethylamino)ethylene (TDAE)). Other difluoroacetyl derivatives 72 were also obtained in good to moderate yields (see table below) starting from 2-chloro-2,2-difluoromethylketones.
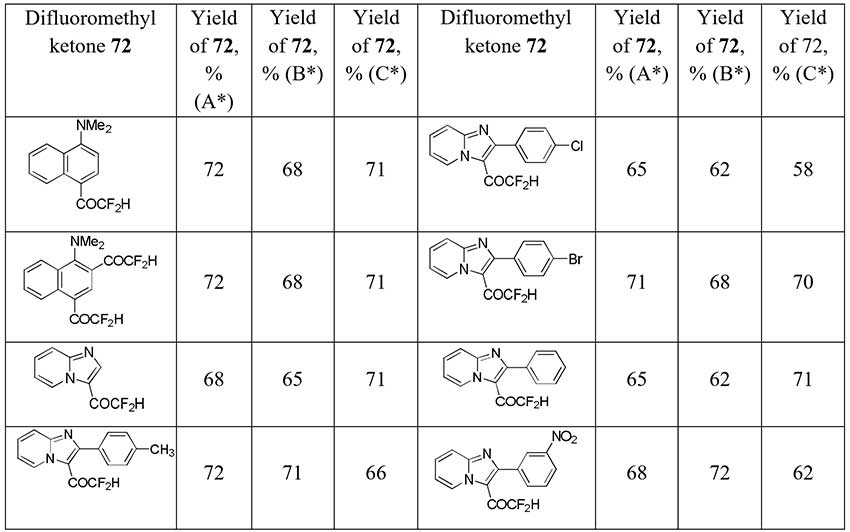
* Method shown in the Scheme 26
The scientists from Turkey [39] have shown that, in contrast to acylphosphonates with an aliphatic substituent, benzoyl phosphonates lead mainly to vinyl phosphonates when reacted with the Ruppert-Prakash reagent. Vinyl phosphonates can be a precursors of difluoromethyl ketones, as was shown by an example of difluoroacetophenone (56) synthesis in high yield.
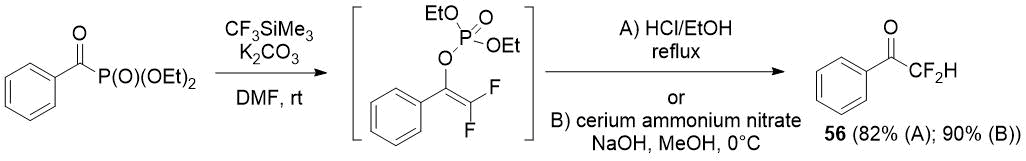
Scheme 27.
Pentafluoroacetone (75) can be obtained from chloropentafluoroacetone (73), available from commercial sources, by a three-step synthesis consisting in the Perkov reaction with the formation of the corresponding vinyl phosphonate 74 and subsequent hydrolysis [40].
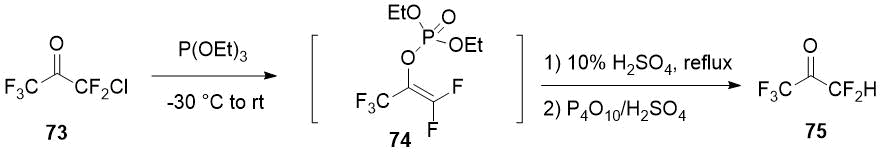
Scheme 28.
Conclusion
There is no universal method for difluoromethyl ketones preparation up to now. All of the reported above methods of synthesis have quite poor substrate scope. An additional activation of the carbonyl substrate is required for selective electrophilic difluorination. In this regard, approaches utilizing a difluoromethylation or difluoroacylation reagents or their equivalents seem interesting and promising. The rapid development of organofluorine chemistry methods and interest in α,α-difluoroketones specifically forces the progress in this field, a steady increase in the number of relevant publications in recent years confirms this fact.
Acknowledgement
This work was supported by Ministry of Science and Higher Education of the Russian Federation.
References
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly, N.A. Meanwell, J. Med. Chem. 2015, 58, 8315-8359.
- E. Camerino, D. M. Wong, F. Tong, F. Körber, A. D. Gross, R. Islam, E. Viayna, J. M. Mutunga, J. Li, M. M. Totrov, J. R. Bloomquist, P. R. Carlier, Bioorg. Med. Chem. Lett. 2015, 25, 4405-4411.
- C. Han, A. E. Salyer, E. H. Kim, X. Jiang, R. E. Jarrard, M. S. Powers, A. M. Kirchhoff, T. K. Salvador, J. A. Chester, G. H. Hockerman, D. A. Colby, J. Med. Chem. 2018, 28, 2697-2700.
- C. B. Kelly, M. A. Mercadante, N. E. Leadbeater, Chem. Comm. 2013, 49, 11133-11148.
- G. Pattison, Eur. J. Org. Chem. 2018, 27-28, 3496-3877.
- W. Wan, J. Hou, H. Jiang, Y. Wang, S. Zhu, H. Deng, J. Hao, Tetrahedron 2009, 65, 4212-4219.
- F. W. van der Mei, C. Qin, R. J. Morrison, A. H. Hoveyda, J. Am. Chem. Soc. 2017, 139, 9053-9065.
- G. A. DeBoos, J. J. Fullbrook, J. M. Percy, Org. Lett. 2001, 3, 2859-2861.
- S. Ge, W. Chaładaj, J. F. Hartwig, J. Am. Chem. Soc. 2014, 136, 4149-4152.
- L. S. Dobsona, G. Pattison, Chem. Comm. 2016, 52, 11116-11119.
- R. J. Linderman, M. S. Lonikar, J. Org. Chem. 1988, 53, 6013.
- J. Ichikawa, T. Sonoda, H. Kobayashi, Tetrahedron Lett. 1989, 30, 5437-5438.
- E. K. Raja, D. A. Klumpp, Tetrahedron Lett. 2011, 52, 5170-5172.
- E. Schmitt, B. Rugeri, A. Panossian, J.-P. Vors, S. Pazenok, F. R. Leroux, Org. Lett. 2015, 17, 4510-4513.
- L. M. Grieco, G. A. Halliday, C. P. Junk, S. R. Lustig, W. J. Marshall, V. A. Petrov, J. Fluor. Chem. 2011, 132, 1198-1206.
- T. L. Andersen, M. W. Frederiksen, K. Domino, T. Skrydstrup, Angew. Chem. Int. Ed. 2016, 55, 1-6.
- T. Brigaud, P. Doussot, C. Portella, J. Chem. Soc., Chem. Commun. 1994, 2117.
- H. Amii, T. Kobayashi, Y. Hatamoto, K. Uneyama, Chem. Comm. 1999, 1323-1324.
- G. K. S. Prakash, J. Hu, G. A. Olah, J. Fluor. Chem. 2001, 112, 355-360.
- K. Boonkitpattarakul, D. Soorukram, P. Tuchinda, V. Reutrakul, M. Pohmakotr, J. Fluor. Chem. 2011, 132, 987-990.
- J. Phetcharawetch, N. M. Betterley, D. Soorukram, M. Pohmakotr, V. Reutrakul, C. Kuhakarn, Eur. J. Org. Chem. 2017, 6840-6850.
- L. Zhang, Y. Li, J. Hu, J. Fluor. Chem. 2007, 128, 755-761.
- J. Chen, J.-H. Lin, J.-C. Xiao, Tetrahedron 2018, 74, 4295-4297.
- S. R. Piettre, C. Girol, C. G. Schelcher, Tetrahedron Lett. 1996, 37, 4711-4712.
- Y. Gu, D. Chang, X. Leng, Y. Gu, Q. Shen, Organometallics 2015, 34, 3065-3071.
- A. L. Trifonov, V. V. Levin, M. I. Struchkova, A. D. Dilman, Org. Lett. 2017, 19, 5304-5307.
- V. V. Levin, V. O. Smirnov, M. I. Struchkova, A. D. Dilman J. Org. Chem. 2015, 80, 9349-9353.
- S. Stavber, M. Zupan, J. Org. Chem. 1987, 52, 5022-5025.
- M. Zupan, J. Iskra, S. Stavber, J. Org. Chem. 1995, 60, 259-260.
- S. Stavber, M. Zupan, Synlett 1996, 7, 693-694.
- W. Ying, D. D. DesMarteau, Y. Gotoh, Tetrahedron 1996, 52, 15-22.
- W. Peng, J. M. Shreeve, J. Org. Chem. 2005, 70, 5760-5763.
- D. J. Leng, C. M. Black, G. Pattison, Org. Biomol. Chem. 2016, 14, 1531-1535.
- Y.-L. Li, J. Li, J. Deng, Adv. Synth. Catal. 2017, 359, 1407-1412.
- Y. Tanaka, T. Ishihara, T. Konno, J. Fluor. Chem. 2012, 137, 99-104.
- P. Peng, J.-J. Wu, J.-Q. Liang, T.-Y. Zhang, J.-W. Huang, F.-H. Wu, RSС Adv. 2017, 7, 56034-56037.
- J.-W. Yuan, S.-N. Liu, W.-P. Mai, Org. Biomol. Chem. 2017, 15, 7654-7659.
- W. R. Dolbier, M. Médebielle, S. Ait-Mohand, Tetrahedron Lett. 2001, 42 4811-4814.
- A. S. Demir, S. Eymur, J. Org. Chem. 2007, 72, 8527-8530.
- B. B. Randolph, D. D. DesMarteau, J. Fluor. Chem. 1993, 64, 129-149.
ARTICLE INFO
Received 04 October 2019
Accepted 16 October 2019
Available online October 2019
Recommended for publication by Prof. S. Igumnov
Fluorine Notes, 2019, 126, 9-10
