Received: November 2019
DOI 10.17677/fn20714807.2019.06.01
Fluorine Notes, 2019, 127, 1-2
SYNTHESIS OF 2-FLUOROOCTANOIC ACID: AN ADVANCED ORGANIC CHEMISTRY LABORATORY EXPERIMENT
Anikó Nemes,* Dénes Szabó, József Rábai
Institute of Chemistry, ELTE Eötvös Loránd University, Budapest 1117, Hungary
E-mail: neagaft@caesar.elte.hu (Dr. Anikó Nemes)
Abstract: A multiple step synthesis of 2-fluorooctanoic acid 4 as individual project in Advanced Organic Chemistry Laboratory course is described. As theoretical background students need to know about enolate chemistry (Hell-Volhard-Zelinsky reaction), nucleophilic substitution and elimination reactions as well. During their practical work, advanced laboratory techniques such as slow addition of reactants, in situ reagent formation and vacuum distillation were introduced. After each step the products were characterized using boiling point or melting point determination and NMR spectroscopy (1H, 13C, 19F).
Keywords: α-fluoroalkanoic acids, potassium fluoride, Hell-Volhard-Zelinsky bromination, substitution, education.
Background
Fluorine-containing organic compounds play important roles in materials science [1] and medicinal / pharmaceutical chemistry [2], thus the synthesis of these types of molecules is an area of great interest [3]. In the last few decades around 20% of the new active pharmaceutical ingredients (APIs) contain at least one fluorine atom [4]. During the drug development process medicinal chemists modify the so-called lead compound to get a drug molecule having the optimal biological activity with optimal pharmacological properties. Replacement of a hydrogen with a fluorine atom can affect the binding to the target protein, the metabolism of the molecule in biological system, and/or the pharmacokinetical properties [5]. Introduction of a fluorine atom to an aromatic ring usually influences the protein-binding through π-π interactions, hydrogen bond affinity and the oxidative metabolism of the molecule; while aliphatic fluorine substitution changes the stable conformation of the small molecule, thus has effect on the protein-binding [6]. For these reasons experience in synthesis of fluorine-containing molecules is important skill for a chemist [7], [8], [9].
In spite of the great importance of organofluorine compounds most organic chemistry textbook omit the theory and synthetic methods of the formation of C-F bonds. Although other carbon-halogen bonds can be formed relatively easily and selectively, formation of C-F bonds is usually not selective and require corrosive reagents and special equipment. The simplest way to synthesize fluorine-containing aliphatic compounds is the displacement of a leaving group by a nucleophilic fluoride usually in a form of KF. The most important disadvantage of these reactions is that usually high degree of elimination side reaction occurs. The optimal yield often accessible by lower degree of conversion, thus the product may contain starting material in addition to elimination products.
2-Fluorocarboxylic acids can be synthesized in three steps starting from the appropriate carboxylic acid as shown in Scheme 1 for the synthesis of 2-fluorooctanoic acid 4. The first step is the halogenation of the carboxylic acids at the α-position (Hell–Volhard–Zelinsky reaction) and the esterification in a one pot reaction. Then the bromine atom of the formed methyl 2-bromocarboxylate 2 is substituted by fluorine using KF in acetamide, and finally the ester group was hydrolyzed to afford the 2-fluorocarboxylic acid.
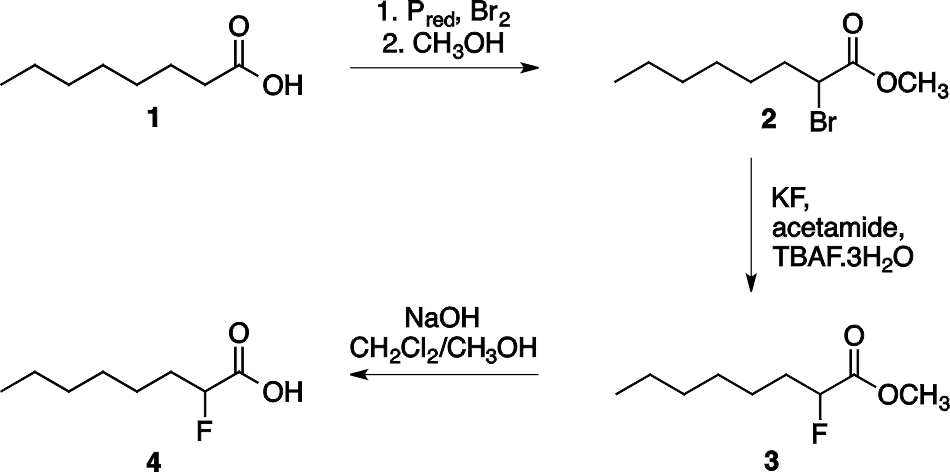
Scheme 1. Synthesis of 2-fluorooctanoic acid 4.
The main teaching goal of this individual laboratory assignment is to show the difficulty of linear synthesis sequences and the importance of the integration of theoretical background into laboratory practice. At the same time this synthesis introduces the students to the use of advanced laboratory techniques (handling highly exothermic reaction, selective reaction facilitated product separation/purification, vacuum distillation) and improves their laboratory skills. These experiments also underline the importance of the monitoring of the individual reactions, moreover the structure and product ratio determination by NMR spectroscopy.
This multistep synthesis was performed by first and second year graduate students, within the framework of Advanced Organic Chemistry Laboratory course, where each student had different individual synthesis projects.
Pedagogical goals and learning outcomes
The synthesis of 2-fluorooctanoic acid was designed to imitate research and development projects in real life. In Advanced Organic Chemistry Laboratory the students received the original research papers of the synthesis of similar molecules like 2-fluorostearic acid [10], 2- fluorohexanoic acid [11] or other long chain derivatives [12] and they must adapt the experimental procedures to their starting materials. The chemical reactivity of these compounds is same, but the physical properties are different (like solubility, melting and boiling point, consistency of the materials, etc.), thus the students need to modify the work- up and purification procedures. This laboratory scale synthesis assignment also can be used in "normal" Organic Chemistry Laboratory, where the instructor gives detailed experimental description of the reactions starting from octanoic acid. In both cases students must use NMR spectroscopy as a powerful analytical technique to determine the product ratio.
Pedagogical Goals
During the synthesis project laboratory skills, integrated thinking skill about the theory of the reaction, TLC and NMR spectroscopy knowledge of the participating students will improve.
- The first reaction step shows the significance of using pure starting materials, and the handling of highly exothermic reactions by slow addition of one of the reagents.
- The second step demonstrates the importance of the knowledge of a reaction mechanism including the potential side reactions and utilizing of the different chemical reactivity of the side product and the necessity of using advanced analytical techniques.
- During the preparation and lab report writing students will reinforce their previous knowledge of the reactivity of carboxylic acids and their derivatives (also the relative reactivity of them), the aliphatic nucleophilic substitution and elimination reactions and basic NMR spectroscopy.
Learning significance
Students will be able to
- work with standard laboratory equipments
- utilize NMR spectroscopy to determine quality and quantity of side products, and determine the structure of the product
- plan a synthesis sequence with time efficiency
- work with hazardous materials, inert gases and equipments under vacuum
- write detailed lab reports with all necessary items (physical properties of the starting materials and products, degree of purity, chromatography details, assignment of NMR spectra).
With the prelab quiz the instructor ensures that the students are familiar with the applied laboratory techniques (use of equipments, such as reactors, rotavap, distillation equipment, vacuum source), and the theory behind the reaction. During the experiment the instructor gives verbal feedback on an individual basis on the performance of the students to guarantee maximal learning outcome.
Experimental overview
The three step synthesis of 2-fluorooctanoic acid 4 was shown in Scheme 1. The bromination of octanoic acid 1 and subsequent esterification provides methyl 2- bromooctanoate 2 as the first intermediate. In the next step, in addition to the halogen exchange an elimination side reaction also takes place to afford an alkene (methyl (E)-2- octenoate, 5). This side product cannot be separated from the desired molecule by distillation since their boiling points are close (94-96 °C / 20 mmHg for 3 and 89-91 °C / 9 mmHg for 5). For the effective separation the reaction mixture was treated with bromine which transformed the alkene to the vicinal dibromide 6. This dibromide has considerably higher boiling point than the desired product, thus they can be separated by vacuum distillation (see Scheme 2).
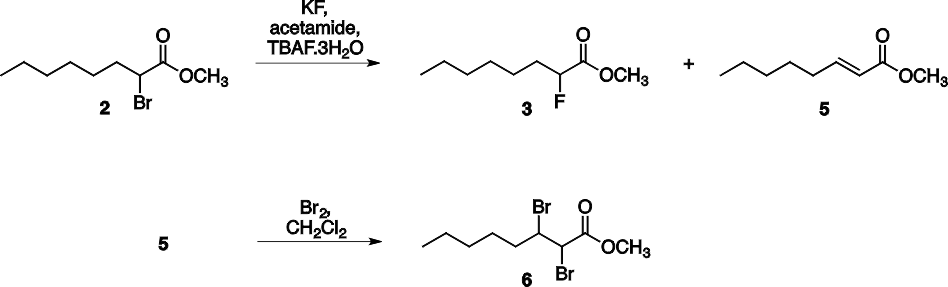
Scheme 2. Reactions of the second synthetic step.
The third step of the reaction is an ester hydrolysis, which results in the end product carboxylic acid 4. Students work individually on the experiments, and after each step they perform 1H and 13C NMR spectroscopy to determine the structure of the product(s) and in the case of the second step the ratio of the side product. The experiment requires about three weeks with each an 8 h laboratory period to complete.
Experiment
The synthesis and purification of the methyl 2-bromooctanoate 2 and the preparation of the substitution reaction was performed in an 8 h lab period (for most students the completion required 7 h). The work up of the substitution reaction and the purification of the methyl 2-fluorooctanoate 3 was accomplished in the second 8 h lab period (for most students the completion required 6 h). The hydrolysis of the above ester and the spectroscopic measurements and structure determination was carried out on the third 8 h lab period (for most students the completion required 8 h). The written reports need to submit in 4 days then feedback should be given during the following laboratory period. A detailed description of the experiment can be found in the Experimental section.
Hazards
All experiments should be performed in a well-ventilated fume hood with appropriate personal protective equipment. Students are required to wear safety goggles and use gloves at all times.
All compounds used for these experiments are classified as irritant. Solvents cause irritation to skin, eyes and respiratory tract, and are possible carcinogenic compounds; furthermore, methanol is also flammable as liquid and vapor. Octanoic acid is very hazardous in case of ingestion, while 2-fluorooctanoic acid is not toxic [13,14]. NaOH and HCl are corrosive. Elemental bromine liquid or spray mist may produce tissue damage particularly on mucous membranes of eyes, mouth and respiratory tract. Skin contact may causes burns. Inhalation of the spray mist may generate severe irritation of respiratory tract, characterized by coughing, choking, or shortness of breath. Severe over–exposure can result in death.
Results and discussion
The preparation of the 2-fluorooctanoic acid from octanoic acid was developed and fully tested by the authors of this article. After the tests the whole synthesis project was performed by 4 M.Sc. students during two semesters. Typical student yield for the three-step sequence was 17 %.
The first bromination could be performed according to general procedure. It is important to emphasize that using pure red phosphorus the reaction is much faster than with technical grade phosphorus. In the former case methanol was added to the reaction mixture after 2 h, while in the latter case the reaction takes place only with 8 h additional stirring. Prior to use red phosphorus was treated with boiling water then filtered and dried. The pure methyl 2- bromooctanoate could be synthesized with an average 89 % yield.
The substitution reaction of 2 was performed using KF as a fluorine source in acetamide, in the presence of TBAF3H2O as catalyst. In order to drive back the unwanted elimination side reaction for the optimal isolated yield the reaction was carried out at 75 °C for 60 h, and the reaction mixture was worked up at about 60 % conversion.
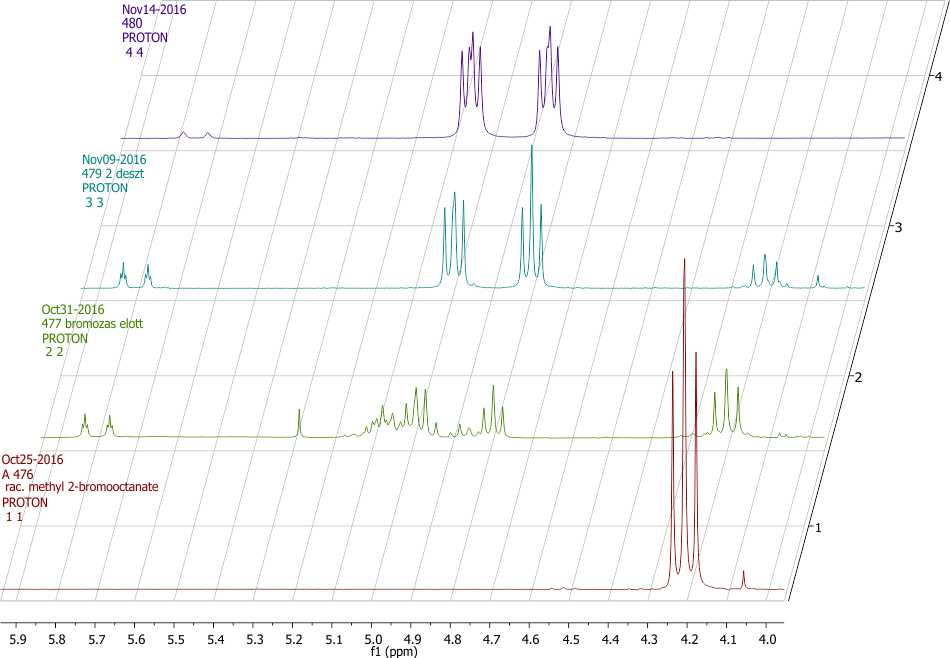
Scheme 3. Partial 1H NMR spectra (4.0-6.0 ppm) of compounds 2 (shown in red, 11), crude 3 (shown in green, 22), pure 3 (shown in blue, 33) and 4 (shown in purple, 44).
Product ratio was determined by 1H NMR spectroscopy using the integral values of the well distinguishable CHCOOCH3 peaks (Scheme 3). Methyne peak of 2 can be found as a doublet of doublets (dd) at 4.21 ppm, while CH peak of 3 shows doublet of doublets of doublets (ddd) multiplicity at 4.90. The alkene side product 5 has the signs of CHCOOCH3 at 5.78 and 5.82. In a typical experiment the crude product after the reaction with KF contained 46 % of substituted product 3, 14 % of 5 and 40 % of starting material 2. (Scheme 2. and 3.)
As mentioned above the crude product was purified in two stages, first the dried CH2Cl2 solution of crude 3 was reacted with elemental bromine, then the obtained mixture was purified by fractional distillation under reduced pressure using Vigreaux column. Typical student yield for this step was 36 %. As shown in Scheme 3., this product also contained some 2 (about 13 %) and 5 (about 8 %), but we found, that using this mixture in the last hydrolysis step the product was pure 2-fluorooctanoic acid 4. Alternatively, the methyl 2- bromooctanoate 2 and methyl 2-fluorooctanoate 3 can be separated also by column chromatography, but this method is time consuming and requires a lot of solvent.
The third and last step of this reaction sequence was the hydrolysis of the ester 3 under basic conditions in CH2Cl2 – CH3OH mixture with a typical student yield of 53 %.
Conclusions
This multistep synthesis served the introduction of several new laboratory techniques that students had previously not performed individually (vacuum distillation), and the application of spectroscopy for determination of molecular structure and product ratio. Students needed to apply wide range of theoretical knowledge during the synthesis. They were also introduced to one of the strategies one can consider, when the byproduct and the product have similar boiling point.
Experimental
NMR spectra (1H, 19F, 13C) were recorded on a Bruker Avance 250 MHz instrument at room temperature (295 ± 2 K) in CDCl3. Chemical shifts (δ) are given in ppm units relatively to the internal standards: TMS (δ = 0.00 for 1H) and CFCl3 (δ = 0.00 for 19F). Melting points were determined on a Boetius micro melting point apparatus and are uncorrected. TLC was performed using Merck SiO2 plates; eluent hexane-CH2Cl2 (1:1 v/v).
Methyl 2-bromooctanoate (2)
The suspension of octanoic acid (20.0 g, 138.5 mmol) and red phosphorus (4.29 g, 138.5 mmol) was heated to 80 °C. Then bromine (26.0 ml, 80.7 g, 505 mmol) was added dropwise under stirring such a rate, that the inner temperature remained below 90 °C (cc. 2 h). The suspension was stirred at 80 °C for an additional 2 h, then cooled to 0 °C and methanol (33 ml) was added during 15 min. The reaction mixture was stirred overnight at room temperature, then boiled for 3h. The phases were separated and the lower phase was washed with CH2Cl2 (50 ml). The combined organic phases were washed with water (50 ml) and dried over Na2SO4. The solution was evaporated and the crude product was purified by distillation in vacuum. Yield: 28.9 g (88%), colorless liquid, bp 122‑124°C/20 mmHg.
1H NMR (CDCl3) δ: 0.87 (3H, t, 3JHH = 6.7 Hz, CH3CH2), 1.2-1.5 (8H, m, CH3CH2CH2CH2CH2), 1.9-2.1 (2H, m, CH2CHBr), 3.76 (3H, s, COOCH3), 4.21 (1H, dd, 3JHH = 7.6 Hz, 3JHH = 7.2 Hz, CHBr).
13C NMR (CDCl3) δ: 14.4 (CH3CH2), 22.8 (CH2-7), 27.6 (CH2-4), 28.8 (CH2-5), 31.8 (CH2-6), 35.3 (CH2-3), 46.1 (CHBr), 53.2 (COOCH3), 170.8 (COOCH3).
Methyl 2-fluorooctanoate (3)
The mixture of methyl 2-bromooctanoate (28.87 g, 122 mmol), acetamide (15 g), KF (12.7 g, 219 mmol) and TBAF×3H2O (2.06 g, 6.53 mmol) was stirred at 75 °C for 60 h under nitrogen atmosphere. After cooling to room temperature water (180 ml) was added to the reaction mixture and extracted with CH2Cl2 (3 × 70 ml). To this solution bromine was added under stirring until color of bromine remained. The combined organic phases were washed with K2S2O5 (2 g) solution in water (50 ml), saturated NaHCO3 (2 × 100 ml) and saturated NaCl (100 ml) solutions and dried over Na2SO4. The solvent was removed under vacuum and the crude product was purified by distillation using Vigreaux column. Yield: 10.2 g (47.4 %), colorless liquid of bp 94-96 °C / 20 mmHg.
1H NMR (CDCl3) δ: 0.87 (3H, t, 3JHH = 6.6 Hz, CH3CH2), 1.2-1.5 (8H, m, CH3CH2CH2CH2CH2), 1.7-2.0 (2H, m, CH2CHF), 3.78 (3H, s, COOCH3), 4.90 (1H, ddd, 3JHH = 5.9 Hz, 3JHH = 6.0 Hz, 3JHF = 49.2 Hz, CHF).
13C NMR (CDCl3) δ: 14.4 (CH3CH2), 22.9 (CH2-7), 24.7 (d, 3JCF = 3 Hz, CH2-4), 29.1 (CH2-5), 31.9 (CH2-6), 32.8 (d, 2JCF = 21 Hz, CH2-3), 52.6 (COOCH3), 89.4 (1JCF = 184 Hz, CHF), 170.9 (d, 2JCF = 24 Hz, COOCH3).
19F NMR (CDCl3) δ: -192.46.
2-Fluorooctanoic acid (4)
To the solution of methyl 2-fluorooctanoate (8.00 g, 45 mmol) in CH2Cl2 (80 ml) was added a solution of NaOH (5.45 g, 136 mmol) in methanol (60 ml). The reaction mixture was stirred overnight; then the solvent was removed under vacuum. The residue was dissolved in water (170 ml) cooled to 0 ºC and acidified with cc HCl (20 ml). The precipitate was filtered and dried in vacuum over P2O5. Yield: 3.93 g (54 %), white waxy solid, mp 51-53 ºC. (Literature [19] mp 51-53 ºC).
1H NMR (CDCl3) δ: 0.87 (3H, t, 3JHH = 6.4 Hz, CH3CH2), 1.2-1.4 (8H, m, CH3CH2CH2CH2CH2), 1.7-1.9 (2H, m, CH2CHF), 4.95 (1H, ddd, 3JHH = 4.8 Hz, 3JHH = 6.7 Hz, 3JHF = 49.2 Hz, CHF), 13.10 (1H, br s, COOH).
13C NMR (CDCl3) δ: 14.2 (CH3CH2), 22.3 (CH2-7), 24.2 (d, 3JCF = 3 Hz, CH2-4), 28.5 (CH2-5), 31.4 (CH2-6), 32.1 (d, 2JCF = 21 Hz, CH2-3), 88.7 (1JCF = 180 Hz, CHF), 170.4 (d, 2JCF = 24 Hz, COOH).
Alternative methods
Alternative, multistep routes, for the synthesis of α-fluoro-carboxylates include opening epoxides [15,16], reacting α-hydroxy-acids with DAST [17] or Ishikawa reagent [18], reacting alkenes with [BrF] agents, prepared in situ from HF and a source of electrophilic bromine [19, 20], or building the target α-fluoro acid by combining two fragments, one of which already contains the fluorine atom [21, 22, 23], as summarized by Rozen and co-workers [24]. In addition, the latter researchers introduced an efficient method for synthesizing branched- and/or unbranched α-fluoro-esters via the reaction of the in situ prepared AcOF-CH3CN electrophilic reagent and the appropriate ketene acetals. This method is fast enough for the preparation of the short-lived 18F-isotope (t1/2 = 109.771 min] labelled target compounds as well [24].
Some of these routes ([19, 22, 23]) allow the convenient synthesis of racemic α-fluoro- octanoic acid since F-building blocks, such as diethyl monofluoromalonate {CHF(CO2C2H5)2} and ethyl fluoroiodoacetate (CHIFCO2C2H5) recently became commercially available. However, the present prices of these fluorinated precursors could be a limiting factor for introducing these novel methods into an Advanced Laboratory Course.
Acknowledgements
This work was completed in the ELTE Institutional Excellence Program (1783-3/2018/FEKUTSRAT), supported by the Hungarian Ministry of Human Capacities.
Literature
- R. Berger, G. Resnati, P. Metrangolo, E. Weber, J. Hulliger, Organic Fluorine Compounds: A Great Opportunity for Enhanced Materials Properties, Chem. Soc. Rev., 2011, 40, 3496– 3508. DOI: 10.1039/C0CS00221F.
- J.-P. Begue, D. Bonnet-Delpon, Bioorganic and Medicinal Chemistry of Fluorine, John Wiley & Sons, Inc., 2008. DOI: 10.1002/9780470281895.
- J. Wang, M. Sánchez-Roselló, J. L. Aceña, C. delPozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok, H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011), Chem. Rev. 114 (2014) 2432- 2506. DOI: 10.1021/cr4002879.
- D. O’Hagan; Fluorine in Healthcare: Organofluorine Containing Blockbuster Drugs, J. Fluorine Chem. 131 (2010) 1071–1081. DOI: 10.1016/j.jfluchem.2010.03.003.
- I. Ojima (Ed.), Fluorine in Medicinal Chemistry and Chemical Biology; Wiley-Blackwell, Chichester, 2009. DOI: 10.1002/9781444312096.
- A. Postigo, Late-Stage Fluorination of Bioactive Molecules and Biologically-Relevant Substrates, Elsevier, 2019.
- L. M. Yagupolskii, Aromatic and Heterocyclic Compounds with Fluorine-Containing Substituents, Naukova Dumka, Kiev,1988.
- G. K. Surya Prakash, F. Wang, Fluorine: The New Kingpin of Drug Discovery, Chim. Oggi/ Chem. Today 30 (2012) 30-36.
- A. Harsanyi, G. Sandford, 2‑Fluoromalonate Esters: Fluoroaliphatic Building Blocks for the Life Sciences, Org. Process Res. Dev. 2014, 18, 981-992. DOI: 10.1021/op500141c
- S. A. Pogány, G. M. Zentner, C. D. Ringeisen, Aqueous Fluoride as a Nucleophile in the Synthesis of 2-Fluorostearic Acid. Synthesis, 1987, 718-719. DOI: 10.1055/s-1987-28058.
- P. Kalaritis, R. W. Regenye, Enantiomerically pure ethyl (R)- and (S)-2-fluorohexanoate by Enzyme-catalyzed Kinetic Resolution. Org. Synth., Coll. Vol. 8, p. 258 (1993). DOI: 10.1002/0471264180.os069.02.
- J. E. Oliver, R. M. Waters, W. R. Lusby, A Convenient Synthesis of α-Fluoro Carboxylic Acids. Synthesis, 1994, 273-275. DOI: 10.1055/s-1994-25457.
- H. Gershon, S. G. Schulman, A. D. Spevack, Organic Fluorine Compounds. III. Action of Perchloryl Fluoride on Substituted Ethyl Cyanoacetates and Animal Toxicities of the Fluorinated Products. J. Med. Chem., 1967, 10, 536-541. DOI: 10.1021/jm00316a008.
- B. C. Saunders, G. J. Stacey, Toxic Fluorine Compounds containing the C-F Link. Part I. Methyl Fluoroacetate and Related Compounds. J. Chem. Soc., 1948, 1773-1779. DOI: 10.1039/jr9480001773.
- H. Keul, B. Pfeffer, K. Griesbaum, Halogenepoxide, 7. Umlagerungsreaktionen bei der Umsetzung von substituierten 2‐Chlor‐ und 2,3‐Dichloroxiranen mit Silbertetrafluoroborat, Chem. Ber. 1984, 117, 2193-2204 https://doi.org/10.1002/cber.19841170613.
- A. O. Amanetoullah, M. M. Chaabouni, A. Baklouti, Synthesis of 2-Fluoro- Acids, Esters, and Amides From α-Dicyanoepoxides, Synth. Commun. 1996, 26, 1155-1161. https://doi.org/10.1080/00397919608003723.
- M. Schlosser, D. Michel, α-Fluoro analogues of inflammation inhibiting α-arylpropionic acids, Tetrahedron, 1996, 52, 8257-8262. https://doi.org/10.1016/0040-4020(96)00411-5.
- S. Watanabe, T. Fujita, M. Sakamoto, H. Endo, Fluorination of aromatic α-hydroxyesters with N,N-diethyl-1,1,2,3,3,3-hexafluoropropaneamine, J. Fluorine Chem. 1990, 47, 187-192. https://doi.org/10.1016/S0022-1139(00)82371-4.
- F. L. M. Pattison, R. L. Buchanan, F. H. Dean, The Synthesis of α-Monofluoroalkanoic Acids, Can. J. Chem. 1965, 43, 1700-1713. https://doi.org/10.1139/v65-224.
- O. Goj, S. Kotila, G. Haufe, Convenient routes to 2-aryl-2-fluoropropionic acids: Synthesis of monofluorinated analogues of (±)-ibuprofen, (±)-naproxen and related compounds, Tetrahedron 1996, 52, 12761-12774. https://doi.org/10.1016/0040- 4020(96)00758-2.
- A. Thenappan, D. J. Burton, Alkylation of (fluorocarbethoxymethylene)tri-n- butylphosphorane: a facile entry to α-fluoroalkanoates, J. Org. Chem. 1990, 55, 2311-2317. https://doi.org/10.1021/jo00295a016.
- Y. Wang, Z. Y. Yang, D. J. Burton, Nickel catalyzed reaction of iodofluoroacetates with alkenes as a facile route to α-fluoroesters, Tetrahedron Lett. 1992, 33, 2137-2140. https://doi.org/10.1016/0040-4039(92)88160-7.
- C. Zhi, Q.-Y. Chen, Novel and practical preparation of a-fluoro-functionalized esters from fluoroiodoacetates, J. Chem. Soc., Perkin Trans. 1, 1996, 1741-1747. DOI: 10.1039/P19960001741.
- S. Rozen, A. Hagooly, R. Harduf, Synthesis of α-Fluorocarboxylates from the Corresponding Acids Using Acetyl Hypofluorite, J. Org. Chem. 2001, 66, 7464-7468. https://doi.org/10.1021/jo010677k
ARTICLE INFO
Received 25 November 2019
Accepted 06 December 2019
Available online December 2019
Recommended for publication by Prof. S. Igumnov
Fluorine Notes, 2019, 127, 1-2
