Received: December 2019
DOI 10.17677/fn20714807.2019.06.02
Fluorine Notes, 2019, 127, 3-4
STEREOCONTROLLED SYNTHESIS OF FLUORINE-CONTAINING FUNCTIONALIZED β-LACTAM DERIVATIVES THROUGH CROSS-METATHESIS REACTIONS
Attila M. Remete,1,2 Zsanett Benke,1,2 Loránd Kiss1,2*
1Institute of Pharmaceutical Chemistry, University of Szeged, H-6720 Szeged, Eötvös u. 6, Hungary
2University of Szeged, Interdisciplinary Excellence Centre, Institute of Pharmaceutical Chemistry, H-6720 Szeged, Eötvös u. 6, Hungary
E-mail: kiss.lorand@pharm.u-szeged.hu
Abstract: Functionalized β-lactams and their derivatives have generated increasing interest in medicinal chemistry over the last decades. Because of their pharmacological potential the chemistry of fluorinated β-lactams and β-amino acids is considered to be an expanding research field. The stereocontrolled synthesis of various fluorine-containing acylic β-lactams has been accomplished from some readily available unsaturated bicyclic β-lactam isomers. The synthetic strategy was based on ring-opening metathesis of unsaturated bicyclic azetidinones followed by cross-metathesis with fluorine-containing olefins. The cross-metathesis transformations, performed under various conditions with the aim of studying chemodiscrimination of the olefin bonds, resulted in the corresponding functionalized β-lactam derivatives.
Keywords: β-lactam, ring opening, functionalization, metathesis, selectivity, fluorine
Introduction
Azetidin-2-ones or β-lactams have received considerable attention in organic and medicinal chemistry. These compounds exhibit a wide range of biological activities. β-Lactam antibiotics are amongst the most important antibacterial agents [1, 2]. Other reported activities include protease inhibition, cholesterol absorption inhibition, human fatty acid synthase inhibition, and antitumor properties [3, 4]. β-Lactams are also valuable synthetic intermediates towards heterocycles, β-amino acids, peptides, taxoids, and other compounds [5-8].
Because of the high importance of fluorinated biomolecules in drug design, the introduction of one or more fluorine atoms into an organic molecule has attracted high interest over the past ten years [9-13]. Thanks to the utility of β-lactams, this interest has also been extended to fluorine-containing azetidin-2-ones. Such compounds are often synthesized by using a fluorinated reactant during heterocyclization [14-19], but late-stage fluorination (fluorine introduction after synthesizing the azetidinone skeleton) can also be applied [20]. β-Lactams expressed their importance in the synthesis of different fluorinated molecules such as sugar–β-amino acid conjugates [15], amido esters [18], aminopropanes and heterocycles [17], as well as taxoids and β-amino acids [20]. Relevant bioactivities were reported for fluoro-taxoids [20] and some N-aryl-3,3-difluoroazetidin-2-ones [21].
One of the most important synthetic developments in the last two decades was the emergence of olefin metathesis reactions. This breakthrough was made possible by commercially available, highly active Ru-based catalysts tolerating varied functional groups (common abbreviations: G1 for Grubbs 1st generation catalysts, G2 for Grubbs 2nd generation catalysts, HG1 for Hoveyda–Grubbs 1st generation catalysts, and HG2 for Hoveyda–Grubbs 2nd generation catalysts) [22, 23]. This powerful, new pathway enabled the synthesis of various compounds, which were previously impossible or hard to prepare [24, 25].
Results and Discussion
In our earlier recent studies, we have successfully transformed unsaturated bicyclic β-lactams into functionalized monocyclic azetidinones via a ring-opening metathesis/cross metathesis (ROM/CM) sequence [26, 27]. Our current aim was to extend this stereocontrolled pathway to the synthesis of fluorine-containing β-lactams utilizing fluorinated olefins in the CM step.
Our first selected substrate was bicyclic lactam (±)-1 derived from 1,5-cyclooctadiene. Its ring opening with ethylene was accomplished according to a literature method [26]. Functionalization of alkenyl-substituted lactam (±)-2 thus obtained was attempted with three different commercially available highly fluorinated alkenes. Cross metathesis reactions in the presence of the four commercial catalysts (G1, G2, HG1, HG2) with 2-bromo-3,3,3-trifluoro-1-propene and 4-bromo-3,3,4,4-tetrafluoro-1-butene failed, but the reaction with allyl 1,1,2,3,3,3-hexafluoropropyl ether proved to be successful. The best yield of dicoupled product (±)-3 was achieved with catalyst HG2 (Scheme 1).
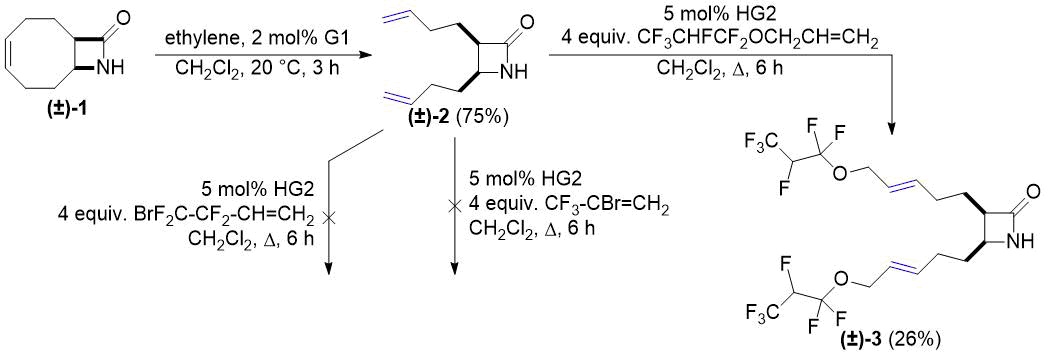
Scheme 1. Cross-metathesis of lactam (±)-2 with fluorinated alkenes.
Compound (±)-4, our next selected substrate, was obtained by N-Boc protection of lactam (±)-1. Modification of the reported procedure [28] resulted in an increased product yield of this step. ROM of compound (±)-4 was performed analogously to ring opening of (±)-1. Product (±)-5 formed in 77% yield was then subjected to CM in the presence of catalyst HG2. Similar to cross metathesis of (±)-2, the reaction succeeded with allyl 1,1,2,3,3,3-hexafluoropropyl ether to afford compound (±)-6, but failed with 2-bromo-3,3,3-trifluoro-1-propene and 4-bromo-3,3,4,4-tetrafluoro-1-butene (Scheme 2).
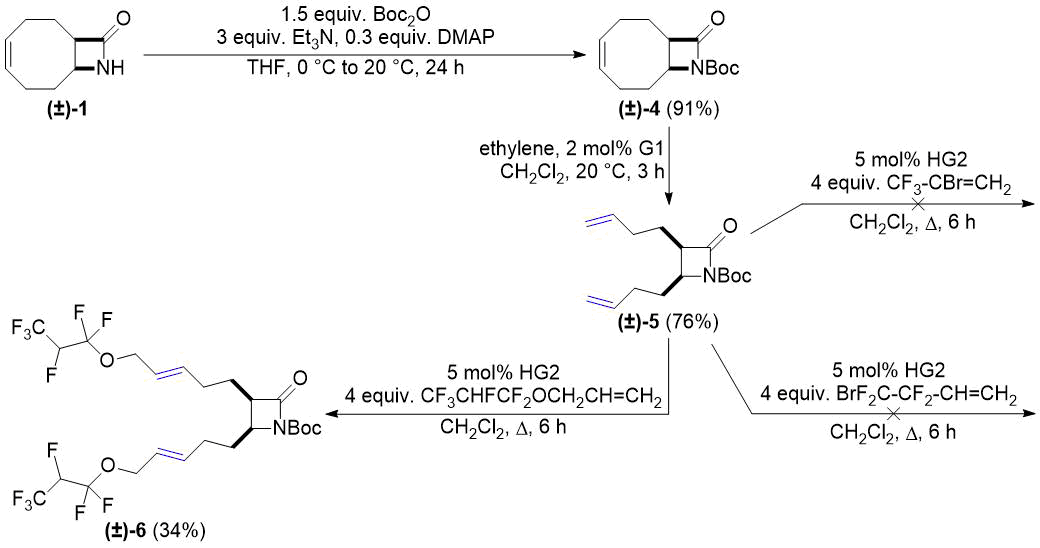
Scheme 2. Cross-metathesis of Boc-protected lactam (±)-5 with fluorinated alkenes.
We continued our work with bicyclic lactam (±)-7 (a regioisomer of lactam (±)-1) derived from 1,3-cyclooctadiene. Its ring opening with ethylene was accomplished according to a literature method described earlier [26]. Taking into account the CM reactions on Scheme 1 and Scheme 2, functionalization of the resulting (±)-8 was attempted only with allyl 1,1,2,3,3,3-hexafluoropropyl ether. The transformation, carried out in the presence of catalyst HG2, gave dicoupled product (±)-9 in a yield of 35% (Scheme 3).
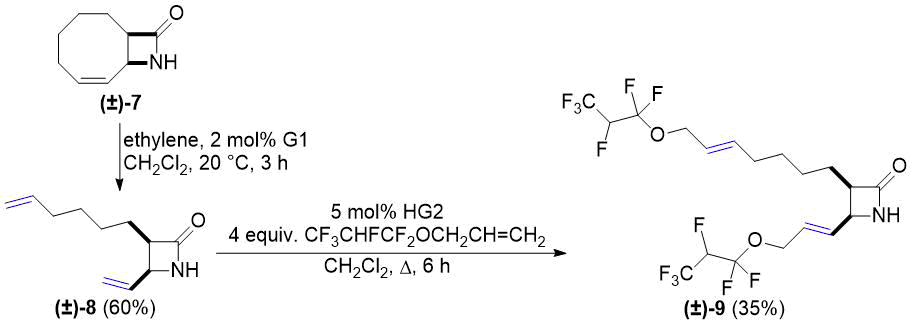
Scheme 3. Cross-metathesis of lactam (±)-8 with fluorinated alkene.
Next, bicyclic lactam (±)-10 (N-Boc-protected derivative of (±)-7) was synthesized. Its reaction with ethylene was performed analogously to ROM of compounds (±)-1 and (±)-4. Cross metathesis of the obtained (±)-11 with allyl 1,1,2,3,3,3-hexafluoropropyl ether produced not only dicoupled product (±)-12 but the formation of monocoupled product (±)-13 was also observed (Scheme 4).
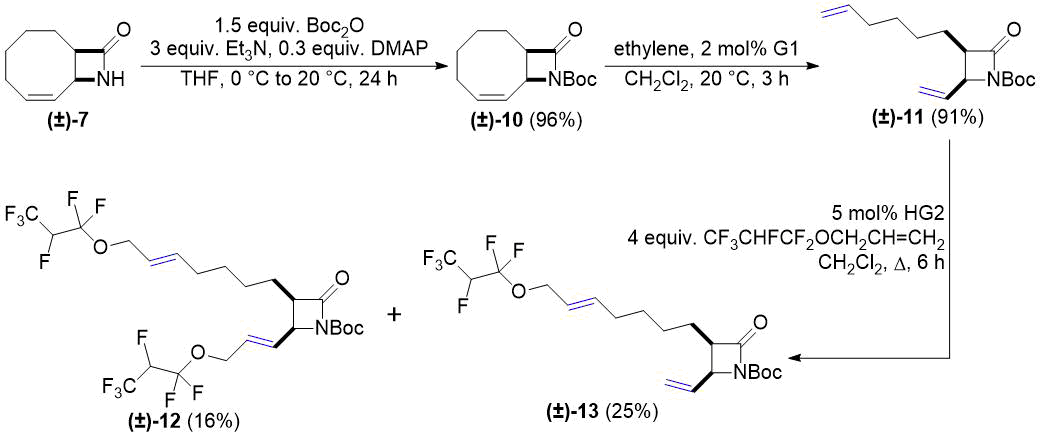
Scheme 4. Cross-metathesis of lactam (±)-11 with fluorinated alkene.
Two factors can contribute to the outcome of this reaction. First of all, chelation of the metallacycle intermediate with carbonyl oxygens is known to hinder further reactions by stabilizing the metallacycle [29-31]. During metathesis of the vinyl group attached directly to the lactam ring of (±)-11, formation of a 6-membered chelate ring with the oxo group of Boc is possible (Scheme 5, (±)-T2 (±)-T3). As a result, reactivity of the C=C bond decreases. Another plausible factor is steric hindrance of this vinyl group attached to the highly substituted azetidinone ring, which slows down its reaction with the Ru-alkylidene catalyst (Scheme 5).
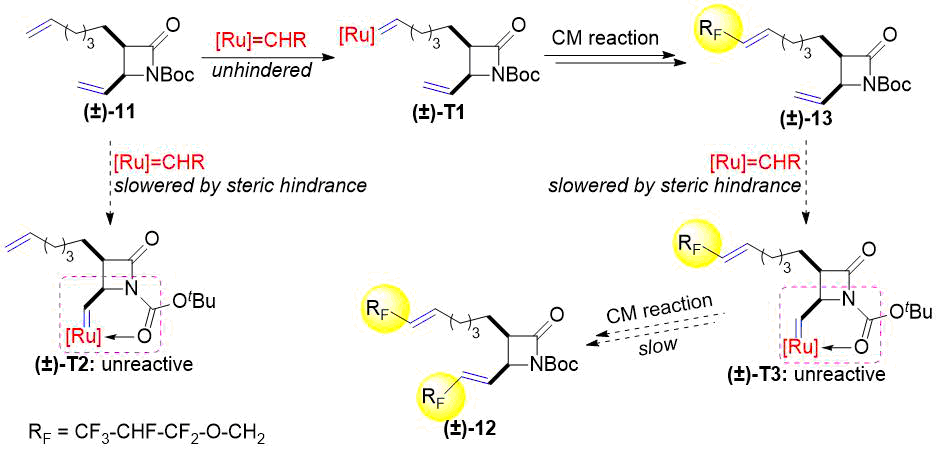
Scheme 5. Formation of mono-metathesized product (±)-13 and decoupled product (±)-12.
In continuation, lactam (±)-14 and its N-Boc-protected derivative (±)-19 were subjected to ROM according to our previous works [26, 27]. Cross metathesis of allyl 1,1,2,3,3,3-hexafluoropropyl ether with compounds (±)-15 and (±)-20 provided temperature-dependent results (Scheme 6 and Scheme 7). Under reflux in CH2Cl2 both (±)-15 and (±)-20 gave the corresponding dicoupled products (±)-16 and (±)-21. At room temperature, in turn, both (±)-15 and (±)-20 were transformed into 1.5:1 mixtures of two monocoupled products. This outcome is similar to our previous results on the chemoselectivity of CM reactions [26, 27]. Unfortunately, separation of the product mixtures in both cases was unsuccessful.
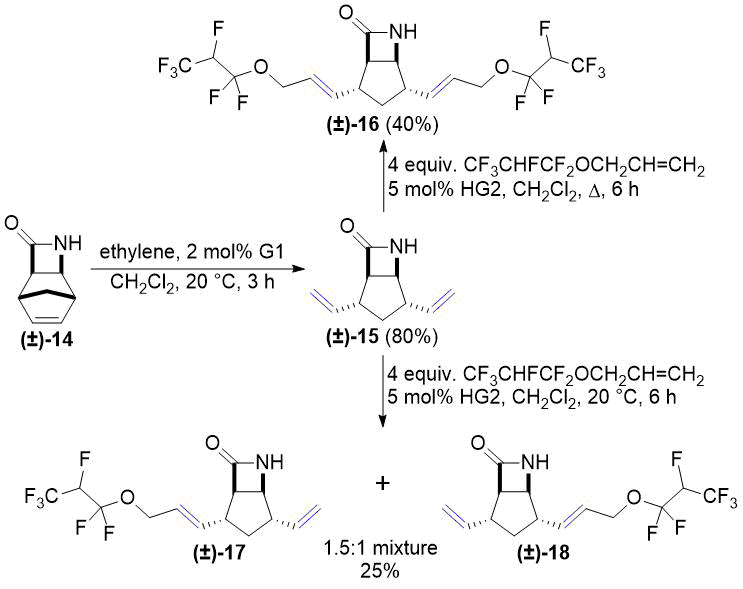
Scheme 6. Cross-metathesis of lactam (±)-15 with fluorinated alkene.
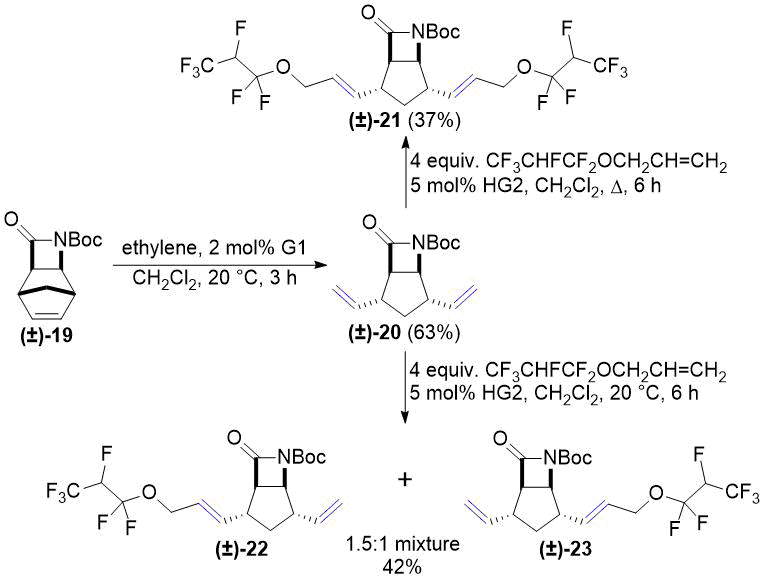
Scheme 7. Cross-metathesis of lactam (±)-20 with fluorinated alkene.
Conclusions
In the current work access to novel β-lactams, possessing fluorine atoms in their substituents, formed under cross-metathesis (CM) reaction conditions has been described. The synthetic concept involved the transformation of various diolefinated β-lactams across CM by using fluorine-containing alkene building elements. CM reaction of the dialkenylated β-lactams, depending on their structure, furnished interesting di- or monocoupled functionalized derivatives. Stereochemical factors through chelate ring formation provide plausibly explanation for the outcome of the CM reactions. Further investigations in view of the synthesis through CM of novel fluorine-functionalized β-lactams are currently being studied in our laboratories.
Experimental
General information
Chemicals were purchased from Sigma-Aldrich. Solvents were used as received from the suppliers. Melting points were determined with a Kofler apparatus. Elemental analyses were performed with a Perkin–Elmer CHNS-2400 Ser II elemental analyser. Silica gel 60 F254 was purchased from Merck. NMR spectra were acquired at room temperature on a Bruker Avance 400 spectrometer (1H frequency 400.13 MHz; 19F frequency 376.50 MHz, 13C frequency 100.76 MHz) or Bruker Avance Neo spectrometer (1H frequency 500.20 MHz; 19F frequency 470.66 MHz, 13C frequency 125.78 MHz) in CDCl3 or D6-DMSO solution, using the deuterium signal of the solvent to lock the field. The 1H and 13C chemical shifts are given relative to TMS and those of 19F to CFCl3 (0.00 ppm).
General Procedure for Ring-Opening Metathesis
β-Lactam (500 mg) was dissolved in anhydrous CH2Cl2 (100 ml) and Grubbs 1st generation catalyst (2 mol%) was added. The mixture was stirred at 20 °C under ethylene for 3 hours monitored by TLC. When the reaction was complete, the solution of NaHCO3 (0.3 g) in H2O (25 ml) and EtOH (5 ml) was added, and the mixture was stirred at 20 °C for another 2 hours with the aim of deactivating the catalyst. Then H2O (30 ml) was added and the mixture was extracted with CH2Cl2 (3×40 ml). The organic layer was dried (Na2SO4) and concentrated under reduced pressure. The crude material was purified by column chromatography on silica gel (n-hexane/EtOAc or n-hexane/acetone).
General Procedure for Cross Metathesis
β-Lactam (150 mg) was dissolved in anhydrous CH2Cl2 (30 ml) and then Hoveyda-Grubbs 2nd generation catalyst (5 mol%) and 3-(1,1,2,3,3,3-hexafluoropropoxy)prop-1-ene (4 equiv.) were added. The mixture was stirred at 40 °C under argon atmosphere for 6 hours monitored by TLC. When the reaction was complete, the solution of NaHCO3 (0.3 g) in H2O (25 ml) and EtOH (5 ml) was added, and the mixture was stirred at 20 °C another 2 hours (catalyst deactivation). After the addition of H2O (20 ml) the mixture was extracted with CH2Cl2 (3×30 ml). The organic layer was dried (Na2SO4) and concentrated under reduced pressure. The crude material was purified by column chromatography on silica gel (n-hexane/EtOAc or n-hexane/acetone).
General Procedure for the Preparation of N-Boc-Protected Lactams
β-Lactam (200 mg) was dissolved in THF (40 ml) then triethylamine (3 equiv.), di-tert-butyl dicarbonate (1.5 equiv.), and 4-dimethylaminopyridine (0.3 equiv.) were added at 0 °C. The mixture was stirred for 1 hour, then it was warmed to room temperature with stirring continued for 24 hours. When TLC indicated complete reaction, H2O (30 ml) was added, and the mixture was extracted with EtOAc (3×20 ml). The organic layer was dried (Na2SO4) and concentrated under reduced pressure. The crude material was purified by column chromatography on silica gel (n-hexane/EtOAc 10:1).
For characterization and NMR data of the new compounds see Supporting Information.
Acknowledgments
We are grateful to the Hungarian Research Foundation (NKFIH No K 119282) for financial support. The financial support of the GINOP-2.3.2-15-2016-00038 project is also acknowledged. This research was supported by the EU-funded Hungarian grant EFOP-3.6.1-16-2016-00008. Ministry of Human Capacities, Hungary grant 20391-3/2018/FEKUSTRAT is also acknowledged.
Supporting Information with 1H and 13C NMR Spectra in PDF version.
References
- Kong, K.-F.; Schneper, L.; Mathee, K. APMIS 2010, 118, 1-36.
- Papp-Wallace, K. M.; Endimiani, A.; Taracila, M. A.; Bonomo, R. A. Antimicrob. Agents Chemother. 2011, 55, 4943-4960.
- Galletti, P.; Giacomini, D. Curr. Med. Chem. 2011, 18, 4265-4283.
- Veinberg, G.; Potorocina, I.; Vorona, M. Curr. Med Chem. 2013, 21, 393-416.
- Kamath, A.; Ojima, I.; Tetrahedron 2012, 68, 10640-10664.
- Piens, N.; De Kimpe, N.; D’hooghe, M. Prog. Heterocycl. Chem. 2016, 28, 27-55.
- Dražić, T.; Roje, M. Chem. Heterocycl. Compd. 2017, 53, 953-962.
- a) Kiss, L.; Fülöp, F. Chem. Rec. 2018, 18, 266-281; b) Kiss, L.; Mándity, I. M.;· Fülöp, F. Amino Acids 2017, 49, 1441–1455.
- Zhou, Y.; Wang, Y.; Gu, Z.; Wang, S.; Zhu, W.; Acena, J. L.; Soloshonok, V. A.; Izawa, K.; Liu, H. Chem. Rev. 2016, 116, 442-518.
- Wang, J.; Sánchez-Roselló, M.; Aceña, J. L.; del Pozo, C.; Sorochinsky, A. E.; Fustero, S.; Soloshonok, V. A.; Liu, H. Chem. Rev. 2014, 114, 2432-2506.
- O’Hagan D. J. Fluorine Chem. 2010, 131, 1071-1081.
- a) Kiss, L.; Forró, E.; Fustero, S.; Fülöp, F. Eur. J. Org. Chem. 2011, 4993-5001; b) Kiss, L.; Remete, A. M. Eur. J. Org. Chem. 2019, 5574–5602.
- Nonn, M.; Kiss, L.; Haukka, M.; Fustero, S.; Fülöp, F. A Org. Lett. 2015, 17, 1074-1077 (and references therein).
- Tarui, A.; Kondo, S.; Sato, K.; Omote, M.; Minami, H.; Miwa, Y.; Ando, A. Tetrahedron 2013, 69, 1559-1565.
- Sundell, R.; Siirola, E.; Kanerva, L. T. Eur. J. Org. Chem. 2014, 6753-6760.
- Trulli, L.; Raglione, V.; Fioravanti, S. Eur. J. Org. Chem. 2018, 3743-3749.
- Thi, H. D.; Thuy, G. L. N.; Catak, S.; Van Speybroeck, V.; Nguyen, T. V.; D’hooghe, M. Synthesis 2018, 50, 1439-1456.
- Thi, H. D.; Goossens, H.; Hertsen, D.; Otte, V.; Nguyen, T. V.; Van Speybroeck, V.; D’hooghe, M. Chem. Asian J. 2018, 13, 421-431.
- El Dine, A. N.; Grée, D.; Roisnel, T.; Caytan, E.; Hachem, A.; Grée, R. Eur. J. Org. Chem. 2016, 556-561.
- Kuznetsova, L.; Ungureanu, I. M.; Pepe, A.; Zanardi, I.; Wu, X.; Ojima, I. J. Fluorine Chem. 2004, 125, 487-500.
- Joyeau, R.; Felk, A.; Guillaume, S.; Wakselman, M.; Vergely, I.; Doucet, C.; Boggetto, N.; Reboud-Ravaux, M. J. Pharm. Pharmacol. 1996, 48, 1218-1230.
- Higman, C. S.; Lummiss, J. A. M.; Fogg, D. E. Angew. Chem. Int. Ed. 2016, 55, 3552-3565.
- Monsaert, S.; Vila, A. L.; Drozdzak, R.; Van Der Voort, P.; Verpoort, F. Chem. Soc. Rev. 2009, 38, 3360-3372.
- Prunet, J.; Eur. J. Org. Chem. 2011, 3634-3647.
- Smith, B. J.; Sulikowski, G. A. Angew. Chem. Int. Ed. 2010, 49, 1599-1602.
- Kardos, M.; Kiss, L.; Fülöp, F. Asian. J. Org. Chem. 2015, 4, 1155-1159.
- Kardos, M.; Kiss, L.; Haukka, M.; Fustero, S.; Fülöp, F. Eur. J. Org. Chem. 2017, 1894-1901.
- Fülöp, F.; Forró, E.; Tóth, G. K. Org. Lett. 2004, 6, 4239-4241.
- BouzBouz, S.; Cossy, J. Org. Lett. 2001, 3, 1451-1454.
- Boufroura, H.; Mauduit, M.; Drège, E.; Joseph, D. J. Org. Chem. 2013, 78, 2346-2354.
- Nonn, M.; Benke, Z.; Fustero, S.; Fülöp, F.; Kiss, L. Eur. J. Org. Chem. 2019, 5285–5293.
ARTICLE INFO
Received 02 December 2019
Accepted 13 December 2019
Available online December 2019
Recommended for publication by Prof. József Rábai
Fluorine Notes, 2019, 127, 3-4
