Received: April 2020
DOI 10.17677/fn20714807.2020.02.03
Fluorine Notes, 2020, 129, 5-6
SYNTHESIS OF N-METHYL-N-POLYFLUOROALKYL-D-GLUCAMINES AND N-METHYL-(FLUOROUS ACID D-GLUCAMIDES)
Norbert Baris, Anikó Nemes, Dénes Szabó, Gitta Schlosser, Antal Csámpai and József Rábai*
Institute of Chemistry, ELTE Eötvös Loránd University, Budapest 1117, Hungary
E-mail:
rabai@elte.hu
Abstract: The selective N-alkylation or N-acylation of D-glucamine using (perfluoroalkyl)propyl iodides/mesylates or fluorinated carboxylic acids allowed the synthesis of novel fluorous chiral resolving agents.
Keywords: fluorous, chiral tertiary amines/amides, carbohydrate based amphiphiles, alkylation, acylation.
Living organisms usually synthesize only one of the two enantiomers of chiral molecules, but in the chemical syntheses often racemic compounds are obtained. The biological activity of enantiomers may be different or even opposite, so enantiomeric separations are necessary and inevitable for the pharmaceutical and fine chemical industries. The strict regulations put in place in the 1990’s on production and marketing chiral drugs made a policy change of pharmaceutical industry [1].
There is high level of commercial interest in chirality and the incentive to produce optically pure materials (i.e. single enantiomers) by methods applicable to at least multigram amounts, and in many cases to hundreds and thousands of tonnes [2].
There are two basic approaches for obtaining chiral compounds: resolution and asymmetric synthesis [3]. Although crystallization is the leading technique for separation of diastereomers [4], several non-conventional techniques are complementary with it or provide the only solutions for special cases [5].
We have disclosed our results on the development of nonconventional optical resolution methods based on the separation of diastereomeric salts or complexes using sulfoxide carboxylic acids as models. Such methods include salting-out selective extraction (‘SOSE’) [6,7] – an early example of today’s enantioselective liquid-liquid extraction – (‘ELLE’) [8], chiroselective transport through liquid membranes [7], and heat-facilitated crystallization (‘HFC’) [7,9,10]. In addition, some (CF3)3CO-substituted fluorous carboxylic acids were synthesized and introduced as new chiral solvating reagents for the determination of enantiomeric ratios of selected amines using 1H and 19F NMR spectroscopy [11].
Carbohydrate based resolving agents play important role in the production of the single enantiomer of active pharmaceutical ingredients [3]. Thus, (S)-(+)- Naproxen was directly resolved from the racemate with high enantiopurity (>95% e.e.) by inclusion crystallization using N-octyl-D-(-)-glucamine as the chiral host [12].
The manufacture of N-polyhydroxyalkyl amines, such as N-methyl D-(-)- glucamine (2), has been known for many years, and such materials are available commercially. The fatty acid amide surfactants (3) are made by the acylation reaction of the unprotected 2O amine (2) with a fatty acid ester (Scheme 1) [13].
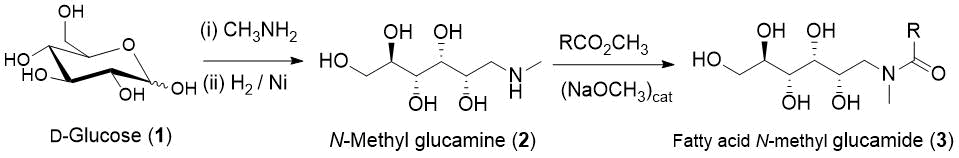
Scheme 1. Manufacturing scheme for the production of fatty acid glucamides.
The title compounds were designed to exploit the unique effects of perfluoroalkyl-groups [14] on the substituted molecules’ microscopic [15] and macroscopic [16] properties. Here we disclose the synthesis of fluorous chiral amines 5a-c and amides 7a-b as shown by Scheme 2 and Scheme 3, respectively.
In the alkylation reactions fluorous alkyl iodides 4a-c [17] were reacted with 2 molar excess of N-methyl D-glucamine (2) and that of K2CO3 in boiling acetonitrile for 4h to achieve full conversion of 4a-c (TLC). Following solvent evaporation the excess of 2 was removed by triturating the crude with water. Then the solid residues were filtered, dried and recrystallized from boiling methanol to afford pure 3O amines 5a-c in medium to good yield as white crystals with narrow melting point range. We note, that C8F17(CH2)3OTs was ineffective for the alkylation of 2 in CH3CN with refluxing for days, while the corresponding mesylate was effective (Scheme 2 and Experimental).
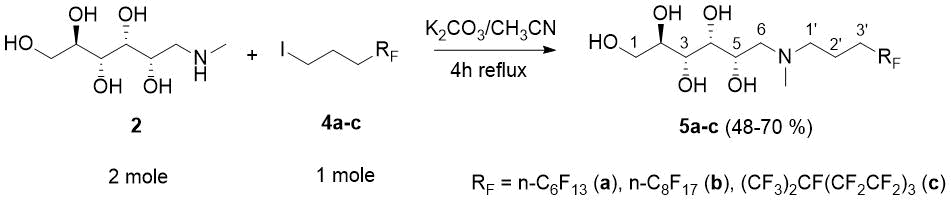
Scheme 2. Preparation of N-methyl-N-[3-(perfluoroalkyl)propyl]-D- glucamines 5a-c. (The numbering presented on 5a-c is used for the assignment of NMR data).
Selective acylation of N-methyl D-glucamine (2) was performed using the mixed-anhydride protocol for the activation of 4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecanoic acid (6a) to afford the fluorous D-glucamide 8a in acceptable isolated yield (Scheme 3.) This carboxyl-activation protocol has been used by Ben et al. [18] for the synthesis of N-methyl-N-octanoyl-D-glucamine (8b).
However, the present fluorous amide 8a gave a massive hydrogel during the aqueous work-up procedure, which was destroyed by the addition of methanol and consecutive boiling to initiate crystallization (Experimental).
We note that the yield of D-glucamide 8b was increased from reported 30% [18] to 79% by the dropwise addition of 2 dissolved in lukewarm methanol to the chilled ether solution of the mixed anhydride 7b (Experimental).
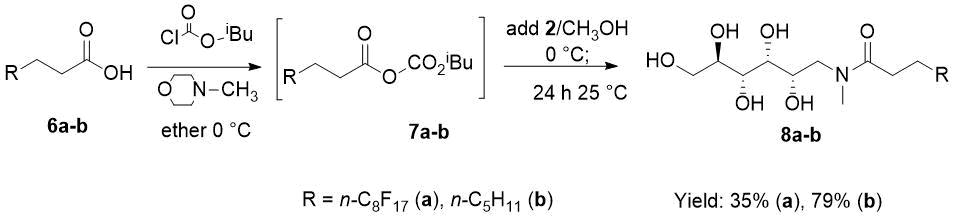
Scheme 3. Selective N-acylation of N-methyl-D-glucamine 2 with in situ generated mixed anhydrides 7a-b.
All new compounds were characterized by 1H NMR, 13C NMR and HRMS spectroscopy (Experimental, Supporting Information). These novel fluorous tertiary amines 5a-c and amides 8a-b will be tested in for the optical resolution of selected volatile racemic fluorinated compounds in our laboratory.
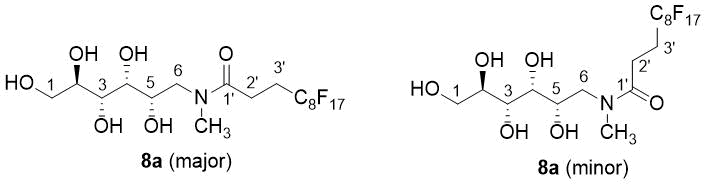
Figure 1. The numbering presented on 8a (major and minor) is used for the assignment of NMR data.
Of note is that the basic tert amine unit in 5a-c seems to accelerate the exchange-processes of the OH protons as their signals merge in the water signal of DMSO-d6 used as solvent for the NMR measurements, while in the 1H NMR spectrum of amide 8a registered in the same solvent, the OH signals are separated as doublets or triplets according to their position in the carbohydrate framework. On the other hand, 8a was present as a ca. 2:1 mixture of two amide rotamers in the sample subjected to NMR measurements (Figure 1). Due to the pronounced deshielding effect of the carbonyl oxygen the 1H-NMR chemical shift of N-CH3 signal of 8a (minor) is markedly downfield-shifted relative to that detected for 8a (major) (3.02 ppm and 2.83 ppm, respectively). Although the fluorinated chain was not assigned in the investigated products, the triplet split of C-3’ resonance clearly refers to the direct attachement of a CF2 group.
Experimental
The fluorinated carboxylic acid 6a was prepared as reported in [19] and [20]. CF3CH2OH and fluorinated precursors were purchased from FC Chemicals, while the other reagents and organic solvents from Sigma-Aldrich and Molar Chemicals Kft (Hungary). The 1H and 13C NMR spectra of all compounds were recorded in DMSO-d6 solution in 5 mm tubes at RT, on a Bruker DRX- 500 spectrometer at 500 (1H) and 125 (13C) MHz, with the deuterium signal of the solvent as the lock and TMS as internal standard. The HSQC, HMBC and COSY spectra, which support exact assignment of 1H- and 13C NMR signals were obtained by using the standard Bruker pulse programs.
Determination of molecular mass and acquisition of the tandem mass spectrum were performed by electrospray ionization mass spectrometry (ESI- MS) on a Bruker Daltonics Esquire 3000 plus (Germany) ion trap mass spectrometer. Melting points were determined on a Boetius micro-melting point apparatus and are uncorrected. The reactions were monitored by TLC (Silica gel 60 F254, Merck Darmstadt). Optical rotations were measured on a Polamat A, Zeiss, Jena polarimeter (concentration c is given as g/100 ml).
(2R,3R,4R,5S)-6-[(methyl(4,4,5,5,6,6,7,7,8,8,9,9,9-tridecafluorononyl)amino]-hexane-1,2,3,4,5-pentanol (5a)
The mixture of N-methyl-glucamine (2, 4.29 g, 22 mmol), 3-(perfluorohexyl)propyl iodide (4a, 5,37 g, 11 mmol) and K2CO3 (3.04 g, 22 mmol) in acetonitrile (180 ml) was boiled for 4h under vigorous stirring. The solvent was removed in vacuo then the residue was stirred with water (40 ml) for 1h, then filtered, washed with water and dried. The crude product was crystallized from MeOH (16 ml) to yield pure 5a (3.75 g, 61.5%), as white crystals with mp 91-93 °C.
[α]578 = – 4.03; [α]546 = – 4.36; [α]436 = – 6.38; [α]406 = –6.71 (c = 3.0 DMF)
1H NMR (DMSO-d6): 3.67 (m, 1H, H-5); 3.58 (m, 1H, H-4); 3.56 (dd, J=10.8 Hz and 3.8 Hz, 1H, H-1A); 3.48 (m, 1H, H-2); 3.43 (dd, J=7.6 Hz and 2.0 Hz, 1H, H-3); 3.38 (dd, J=10.8 Hz and 5.8 Hz, 1H, H-1B); 2.44-2.39 (overlapping m’s, 3H, H-1’ and H-6A); 2.33 (dd, J=13.2 Hz and 6.8 Hz, 1H, H-6B); 2.20 (m, 2H, H-3’); 2.16 (s, 3H, N-CH3); 1.63 (qui, J=7.9 Hz, 2H, H-2’).
13C NMR (DMSO-d6) (non-fluorinated carbons: 72.9 (C-3); 72.2 (C-2); 70.9 (C-4); 70.8 (C-5); 64.0 (C-1); 60.7 (C-6); 56.9 (C-1’); 42.6 (N-CH3);28.4 (t, J=21.2 Hz, C-3’); 18.2 (C-2’).
HRMS (ESI) m/z: calcd. for C16H22F13NO5555.12904; found 555.12721. Mass error: –3.3 ppm.
(2R,3R,4R,5S)-6-[(4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoroundecyl)(methyl)amino]hexane-1,2,3,4,5-pentanol (5b)
Method A:
The mixture of N-methyl-glucamine (2, 5.85 g, 30 mmol), 3-(perfluorooctyl)propyl iodide (4b, 8.85 g, 15 mmol) and K2CO3 (4.14 g, 30 mmol) in acetonitrile (300 ml) was boiled for 4h under vigorous stirring. The solvent was removed in vacuo then the residue was stirred with water (50 ml) for 1h, then filtered, washed with water and dried. The crude product was crystallized from MeOH (35 ml) to yield pure 5b as white crystals (Fw = 655, 6.33 g, 64.5%), mp 103-105°C. Using 30 mmol of 4b 13.74 g (70%) of 5b was obtained as white crystals with mp 104-107°C.
[α]578 = − 3.07; [α]546 = − 3.41; [α]436 = − 6.48; [α]406 = − 6.83 (c = 3, DMF).
1H NMR (DMSO-d6): 3.63 (m, 1H, H-5); 3.54 (m, 1H, H-4); 3.52 (dd, J=10.8 Hz and 3.8 Hz, 1H, H-1A); 3.43 (m, 1H, H-2); 3.36 (dd, J=7.6 Hz and 2.0 Hz, 1H, H-3); 3.32 (dd, J=10.8 Hz and 5.8 Hz, 1H, H-1B); 2.42-2.33 (overlapping m’s, 3H, H-1’ and H-6A); 2.27 (dd, J=13.2 Hz and 6.8 Hz, 1H, H-6B); 2.16 (m, 2H, H-3’); 2.12 (s, 3H, N-CH3); 1.58 (qui, J=7.9 Hz, 2H, H-2’).
13C NMR (DMSO-d6) (non-fluorinated carbons: 72.6 (C-3); 71.9 (C-2); 70.7 (C-5); 70.6 (C-4); 63.9 (C-1); 60.6 (C-6); 56.8 (C-1’); 42.6 (N-CH3); 28.1 (t, J=21.5 Hz, C-3’); 18.0 (C-2’).
HRMS (ESI) m/z: calcd. for C18H22F17NO5 655.12265; found 655.12024. Mass error: -3.7 ppm.
Method B:
A mixture of 3-(perfluorooctyl)propyl mesylate (0.56 g, 1.0 mmol), N-methyl-glucamine (2, 0.39 g, 2 mmol) and K2CO3 (0.28 g, 2 mmol) in acetonitrile (10 mL) eddig a ml kis l volt was stirred and heated at 70oC temperature for 24 h in a sealed vile. Then the solvent was removed by evaporation in vacuum using a rotavap. The solid residue was treated with water (15 ml) and filtered. The crude product was filtered by suction and dried to afford 0.40 g (62%) white crystals with mp 102-104 °C.
HRMS (ESI) m/z: calcd. for C18H22F17NO5 655.12265; found 655.12028. Mass error: -3.6 ppm.
(2R,3R,4R,5S)-6-{[4,4,5,5,6,6,7,7,8,8,9,9,10,11,11,11-hexadecafluoro-10-(trifluoromethyl)undecyl](methyl)amino}hexane-1,2,3,4,5-pentaol (5c)
The mixture of N-methyl-glucamine (2, 7.0 g, 36 mmol), 3-(iso-perfluorononyl)propyl iodide (4c, 11.6 g, 18 mmol) and K2CO3 (4.98 g, 36 mmol) in acetonitrile (350 ml) was boiled for 4h under vigorous stirring. The solvent was removed in vacuo then the residue was stirred with water (60 ml) for 1h. To this aqueous suspension MeOH (130 ml) was added and the mixture was heated to give a clear solution. After cooling the precipitate was filtered, dried and crystallized from MeOH (45 ml) to yield pure 5c (6.1 g, 48%); mp 86-91°C.
[α]578 = −3.07; [α]546 = −3.41; [α]436 = −6.48; [α]406 = −6.83. (c = 1, CF3CH2OH).
1H NMR (DMSO-d6): 3.67 (m, 1H, H-5); 3.58 (m, 1H, H-4); 3.56 (dd, J=10.8 Hz and 3.8 Hz, 1H, H-1A); 3.47 (m, 1H, H-2); 3.40 (dd, J=7.6 Hz and 2.0 Hz, 1H, H-3); 3.35 (dd, J=10.8 Hz and 5.8 Hz, 1H, H-1B); 2.46-2.37 (overlapping m’s, 3H, H-1’ and H-6A); 2.31 (dd, J=13.2 Hz and 6.8 Hz, 1H, H-6B); 2.17 (m, 2H, H-3’); 2.15 (s, 3H, N-CH3); 1.62 (qui, J=7.9 Hz, 2H, H-2’).
13C NMR (DMSO-d6) (non-fluorinated carbons: 72.7 (C-3); 71.9 (C-2); 70.7 (C-4); 70.6 (C-5); 63.9 (C-1); 60.6 (C-6); 56.8 (C-1’); 42.6 (N-CH3); 28.1 (t, J=21.5 Hz, C-3’); 18.0 (C-2’).
HRMS (ESI) m/z: calcd. for C19H22F19NO5 705.11946; found 705.11733. Mass error: -3.0 ppm.
4,4,5,5,6,6,7,7,8,8,9,9,10,10,11,11,11-heptadecafluoro-N-methyl-N-[(2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl]undecanamide (8a)
The solution of 3-perfluorooctyl-propanoic acid 6a (4.92 g, 10 mmol) in diethyl ether (20 ml) was cooled to 0 °C and isobutyl chloroformate (1.37 g, 10 mmol) was added. After stirring for 10 min N-methyl-morpholin (1.01 g, 10 mmol) was added and stirred for another 10 min. The precipitated hydrochloride salt was removed by filtration, then to the filtrate a warm solution (50°C) of N-methylglucamine (0.88 g, 4.5 mmol) in MeOH (30 ml) was dropped during 10 min. The mixture was strirred for a night at room temperature, then the solvent was removed in vacuo. The residue was triturated with diethyl ether, and the crystals were filtered with suction and dried to give (2.6 g) crude. The crude product was crystallized from MeOH (10 ml) to yield pure 8a 1.04 g (35%), mp 95-210 °C. This unique melting behaviour could be the result of changes in the structure of the H-bonded network in the solid state upon heating. It is worth to note that in solution (DMSO-d6) the existence of two distinct rotamers are identified by NMR (cf. Supporting Information).
1H NMR (DMSO-d6): the most diagnostic and unambiguous assignments without providing intensity for 8a (major): 4.94 (d, J=5.0 Hz, 5-OH); 4.34 (t, J=5.5 Hz, 1-OH); 3.77 (m, H-5); 3.56 (m, H-1A);3.37 (m, H-1B); 3.42-3.27 (overlapping m’s, H-6A and H-6B in the major and minor components); 2.83 (s, N-CH3); 2.62 (m, H-3’).
13C NMR (DMSO-d6) the most diagnostic and unambiguous assignments of the non-fluorinated carbons intensity for 8a (major): 170.16 (C-1’); 70.5 (C-5); 63.78 (C-1); 52.3 (C-6);34.1 (N-CH3); 26.5 (t, J=21.3 Hz, C-3’); 23.8 (C-2’).
1H-NMR (DMSO- d6): the most diagnostic and unambiguous assignments without providing intensity for 8a (minor): 4.69 (d, J=5.0 Hz, 5-OH); 4.30 (t, J=5.5 Hz, 1-OH); 3.42-3.27 (overlapping m’s, H-6A and H-6B in the major and minor components); 3.02 (s, N-CH3); 2.80 (m, H-2’).
13C-NMR (DMSO-d6) the most diagnostic and unambiguous assignments of the non-fluorinated carbons intensity for 8a (minor): 170.06 (C-1’); 71.5 (C-5); 63.75 (C-1); 51.6 (C-6); 36.9 (N-CH3); 26.3 (t, J=21.9 Hz, C-3’); 24.2 (C-2’). HRMS (ESI) m/z: calcd. for C18H20F17NO6 669.10192; found 669.10150. Mass error: -6.0 ppm.
HRMS (ESI) m/z: calcd. for C18H20F17NO6 669.10192; found 669.10150. Mass error: -6.0 ppm.
N-methyl-N-((2S,3R,4R,5R)-2,3,4,5,6-pentahydroxyhexyl)octanamide (8b)
To the ice-cold solution of octanoic acid (2.88 g, 20 mmol) in ether (30 ml) ClCOOiBu (2.73g, 20 mmol) was added and the mixture stirred for 10 min. After the addition of N-methylmorpholine (2.02 g, 20 mmol) the precipitated hydrochloride salt was removed by filtration. The filtrate was cooled with ice while a solution of N-methyl-glucamine (1.75 g, 9 mmol) in lookwarm (40-50 °C) ether (60 ml) was added drop-by-drop. Then the mixture was allowed to warm up to room temperature and kept there overnight. The solvent was evaporateted and the residual oil was treated with a small volume of ether and scrubbed to initiate crystallization. Fitration gave 2.60 g (81%) solid, which on recrystallization from acetonitrile (10 ml) gave 2.28 g (79%) white crystals with mp = 84-85 °C.
MS (EI) m/z: calcd for C15H31NO6 321.2; observed 322.4 (M+H)+.
Acknowledgement
The authors thank the National Research, Development and Innovation Office (K115764) for supporting this work. G. S. acknowledges the support by the MTA János Bolyai Research Scholarship and by the MTA Premium Post-Doctorate Research Program of the Hungarian Academy of Sciences (HAS, MTA).
See PDF version for supporting Information with NMR spectra.
Literature
- E. Pálovics, Zs. Szeleczky, B. Fődi, F. Faigl, E. Fogassy, Prediction of the efficiency of diastereoisomer separation on the basis of the behavior of enantiomeric mixtures, RSC Adv., 2014, 4, 21254-21261. DOI: 10.1039/C4RA00526K
- N. A. Vaidya, Diastereomeric crystallisation – the ‘classical’ chiral technology; Innovations in Pharmaceutical Technology (2000) 82-85; accessed at 01 December, 2019. http://iptonline.com/articles/public/IPTNINE82NoPrint.pdf
- D. J. Ager (Ed.), Handbook of chiral chemicals. 2nd ed. ©, 2006, Taylor & Francis Group, https://doi.org/10.1201/9781420027303
- Pálovics, E.; Faigl, F., Fogassy, E., Chap.1. Separation of the Mixtures of Chiral Compounds by Crystallization. Advances in Crystallization Processes, Yitzhak Mastai (Ed.), InTech, 2012. DOI: 10.5772/33592
- E. Fogassy, M. Nógrádi, E. Pálovics, J. Schindler, Resolution of Enantiomers by Non-Conventional Methods. Synthesis, 2005, 1555-1568. DOI: 10.1055/s-2005-869903
- J. Rábai, Salting Out Selective Extraction – A Novel Method for the Optical Resolution of Chiral Sulfinylcarboxylic Acids and Its Application for the Convenient Determination of Optical Purity, Angew. Chem., Int. Ed., 1992, 31, 1631-1633. https://doi.org/10.1002/anie.199216311
- A. Nemes, D. Szabó, J. Rábai, Comparison of resolution methods for racemic 8‑(phenylsulfinyl)-1-naphthoic acid, Tetrahedron: Asymmetry, 2017, 1078-1082. DOI: 10.1016/j.tetasy.2017.07.001
- B. Schuur, B. J. V. Verkuijl, A. J. Minnaard, J. G. de Vries, H. J. Heeres and B. L. Feringa, Chiral separation by enantioselective liquid–liquid extraction, Org. Biomol. Chem., 2011, 9, 36-51. DOI: 10.1039/C0OB00610F
- D. Szabó, A. Nemes, I. Kövesdi, V. Farkas, M. Hollósi, J. Rábai, Synthesis and characterization of fluorous (S)- and (R)-1-phenylethylamines that effect heat facilitated resolution of (±)-2-(8-carboxy-1-naphthylsulfinyl)benzoic acid via diastereomeric salt formation and study of their circular dichroism, J. Fluorine Chem., 2006, 127, 1405-1414. https://doi.org/10.1016/j.jfluchem.2006.05.011
- A. Nemes, E. Vass, I. Jalsovszky, D. Szabó, Synthesis of enantiopure 2‑iodomandelic acid and determination of its absolute configuration by VCD spectroscopy, Chem. Pap., 2019, 73, 47-54. https://doi.org/10.1007/s11696- 018-0568-6
- A. Nemes, T. Csóka, Sz. Béni, V. Farkas, J. Rábai, D. Szabó, Chiral recognition studies of α-(nonafluoro-tert- butoxy)carboxylic acids by NMR spectroscopy, J. Org. Chem., 2015, 80(12), 6267-6274. https://doi.org/10.1021/acs.joc.5b00706
- Xuejun Yuan, Jiguo Li, Yunqi Tian, Gene-Hsiang Lee, Xie-Ming Peng, Rongguang Zhu and Xiaozeng Youa, Efficient resolution of naproxen by inclusion crystallization with N-octyl-glucamine and structure characterization of the inclusion complex, Tetrahedron: Asymmetry, 2001, 12, 3015–3018. https://doi.org/10.1016/S0957- 4166(01)00536-5
- (a) J. J. Scheibel, D. S. Connor, R. E. Shumate, and J. C. T. R. B. St. Laurent, Process for Preparing N-Alkyl Polyhydroxy Amines and Fatty Acid Amides Therefrom in Hydroxy Solvents, EP 0558515B, 1990, Procter & Gamble, Chem. Abstr. 1992, 117, 114045; (b) K. Hill and C. LeHen-Ferrenbach, Sugar-Based Surfactants for Consumer Products and Technical Applications, Chapter 1, In: Cristóbal Carnero Ruiz (Editor), Sugar-Based Surfactants: Fundamentals and Applications, CRC Press Tayrol & Francis Group, Boca Raton, FL, 2009.
- (a) I. T. Horváth, J. Rábai, Facile Catalyst Separation without Water: Fluorous Biphase Hydroformylation of Olefins, Science, 1994, 266, 72-75, DOI:10.1126/science.266.5182.72; (b) Handbook of Fluorous Chemistry, J. A. Gladysz, D. P. Curran, I. T. Horváth (Editors), Wiley/VCH: Weinheim, 2004, DOI: 10.1002/3527603905; (c) Fluorous Chemistry, Volume Editor: I. T. Horváth, Topics in Currant Chemistry, Springer, 2012, 308, Heidelberg. DOI 10.1007/978-3-642-25234-1; (d) J.-M. Vincent, M. Contel, G. Pozzi, R. H. Fish, How the Horváth paradigm, Fluorous Biphasic Catalysis, affected oxidation chemistry: Successes, challenges, and a sustainable future, Coord. Chem. Rev., 2019, 380, 584-599. https://doi.org/10.1016/j.ccr.2018.11.004
- M. Cametti, B. Crousse, P. Metrangolo, R. Milani, G. Resnati, The fluorous effect in biomolecular applications, Chem. Soc. Rev., 2012, 41, 31–42. DOI: 10.1039/C1CS15084G
- M. P. Kraft, Fluorocarbons and fluorinated amphiphiles in drug delivery and biomedical research, Adv. Drug Delivery Rev., 2001, 47, 209-228. DOI: 10.1016/s0169-409x(01)00107-7
- B. Menczinger, G. Jakab, D. Szabó and J. Rábai, Synthesis of 1-iodo-3-perfluoroalkylpropanes and 1-iodo-4- perfluoroalkylbutanes, Fluorine notes, 2014, 94, 7-8. http://notes.fluorine1.ru/public/pdfs/94_4.pdf
- C. J. Capicciotti, M. Leclère, F. A. Perras, D. L. Bryce, H. Paulin, J. Harden, Y. Liu, R. N. Ben, Potent Inhibition of Ice Recrystallization by Low Molecular Weight Carbohydrate-Based Surfactants and Hydrogelators, Chem. Sci., 2012, 3, 1408-1416. DOI: 10.1039/c2sc00885h.
- T. Miura, Y. Hirose, M. Ohmae, and T. Inazu, Fluorous Oligosaccharide Synthesis Using a Novel Fluorous Protective Group, Org. Lett., 2001, 3, 3947-3950. DOI: 10.1021/ol016838o.
- B. Hungerhoff, H. Sonnenschein, F. Theil, Combining Lipase-Catalyzed Enantiomer-Selective Acylation with Fluorous Phase Labeling: A New Method for the Resolution of Racemic Alcohols, J. Org. Chem., 2002, 67, 1781-1785. https://doi.org/10.1021/jo010767p
ARTICLE INFO
Received 01 April 2020
Accepted 08 April 2020
Available online April 2020
Recommended for publication by Prof. Sergey Igoumnov
Fluorine Notes, 2020, 129, 5-6
