Received: October 2021
DOI 10.17677/fn20714807.2021.06.01
Fluorine Notes, 2021, 139, 1-2
SYNTHESIS OF LITHIUM (PERFLUOROALKYL)TRIHYDROALUMINATES Li[RFAlH3]
A.A. Tyutyunova, А.S. Peregudova, S.М. Igumnova,b
aA.N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences,
28 Vavilova St., 119991 Moscow, Russian Federation
bP&M-Invest Scientific Production Association,
28 Vavilova St., 119991 Moscow, Russian Federation
e-mail: tuytuynov@rambler.ru
Abstract: (Perfluoroalkyl)trimethylsilanes were shown to react with LiAlH4 in ethereal solvents to give lithium (perfluoroalkyl)trihydroaluminates Li[RFAlH3], which, when RF = C2F5 and n-C3F7, can be isolated as stable complexes solvated with ethereal solvents, for example Li[C2F5AlH3]2DME, Li[nC3F7AlH3]2DME.
Key words: lithium aluminum hydride, (perfluoroalkyl)trimethyl silanes, lithium (perfluoroalkyl)trihydroaluminates
We have previously shown that the reaction of NaBH4 with CF3SiMe3 in diglyme does not produce Na[CF3BH3], and instead, gives HCF2SiMe3 as a major product [1]. We also failed to prepare (trifluoromethyl)borohydrides by the action of NaBH4 or BH3 (obtained in situ by the reaction of NaBH4 with ClSiMe3) in a diglyme solution on CF3B(OMe)2 or K[CF3B(OMe)3]. The resulting products proved to be unstable and decomposed during isolation, in some cases with strong explosions. The recent report on the synthesis of (perfluoroalkyl)borohydrides indirectly supports our results, and it shows that more stable potassium and cesium (pentafluoroethyl)borohydrides, obtained in pure forms, explode upon friction [2].
A natural question that arose when studying the reaction of RFSiMe3 with NaBH4, is how a similar reaction will proceed with LiAlH4? We have shown that RFSiMe3 reacts with LiAlH4 in ethereal solvents to furnish Li[RFAlH3] and HSiMe3 [3]. In the present paper, we present the results of these studies.
It turned out, that following the addition of CF3SiMe3 (1.1 equiv.) to a solution of LiAlH4 (1 equiv.) in diglyme at room temperature, a highly exothermic reaction is observed after a few minutes. When carrying out this reaction while controlling the temperature of the reaction mixture at ~20°C for 5.5 hours, analysis of the reaction mixture by 1H NMR confirmed the formation of HSiMe3 (1H NMR, δ: 0.56 (d, 9H, 3JHH = 5 Hz, SiMe3), 4.44 (dec, 1H, HSi); 29Si {1H} NMR,δ: -16 (s)), the conversion of CF3SiMe3 is 58% (in 24 h, the conversion of CF3SiMe3 is 79%). The 19F NMR spectrum of the reaction mixture contains a broad peak with a maximum at -175 ppm, indicating the formation of aluminum fluoride complexes [4] (e.g., in [nBu4N][Me2AlF2] and Li[(Me3Si)3CAlF3]THF, the fluorine atoms resonate at -154 [5] and -169 ppm [6], respectively), and also a residual signal of unreacted CF3SiMe3 at -67 ppm. This is consistent with the data of the 29Si{1H} NMR spectrum, which shows the peak corresponding to CF3SiMe3 at δ=5 (q, 2JSiF = 37 Hz, CF3Si). Taking into account that the 1H NMR spectrum of the reaction mixture contains several broad upfield signals (δ: -0.7 and -0.75 ppm), it can be assumed that in this reaction, in addition to HSiMe3, lithium aluminum hydrofluoride complexes are formed. No characteristic peaks appear in the 7Li and 27Al NMR spectra. The reaction in THF provides a similar result. The degree of conversion of CF3SiMe3 to HSiMe3 is 36% (8 h), 55% (32 h), and 63% (56 h); also, lithium aluminum hydrofluoride complexes are formed having similar spectral characteristics. The removal of THF in vacuo, together with the volatile components of the mixture (HSiMe3 and unreacted CF3SiMe3), provides a white solid, which actively reacts with water and methanol. The mass of the residue, as well as the low fluorine content (5%), established by elemental analysis, suggests that this product is Li[AlFH3], which contains unreacted LiAlH4 and solvating THF, probably resulting from the decomposition of the initially formed Li[CF3AlH3]. Purification and identification of lithium aluminum hydrofluoride complexes formed in this reaction was not performed.
In contrast, C2F5SiMe3 reacts with LiAlH4 in diglyme virtually without heat evolution. According to 1H and 29Si{1H} NMR data, the reaction provides HSiMe3, and in the 19F NMR spectrum, in addition to peaks for the starting C2F5SiMe3 (δ: -132 (s, 2F, CF2), -82 (s, 3F, CF3); 29Si {1H} NMR δ: 7.8 (t, 2JSiF = 27 Hz, CF2Si)), signals for the product bearing C2F5 moiety (δ: -126 (br. s, 2F, CF2Al), -84 (s, 3F, CF3)) are observed, where this product is, as we hypothesize, an ate complex of Li[C2F5AlH3], which is consistent with the data of the 27Al NMR spectra, showing the peak at 116 ppm (br.d, CF2Al). However, based on the data of 1H, 19F, 27Al, and 29Si NMR spectra, it turned out that C2F5SiMe3 (1.1 equiv.) reacts with c LiAlH4 (1 equiv.) in diglyme at ~20°C within several days with incomplete conversion of ~70%. This circumstance significantly complicates the isolation of the Li[C2F5AlH3] complex in pure form, due to the presence of unreacted LiAlH4 in the solution. This is confirmed by the 27Al NMR spectrum containing a peak at 102 ppm, br.s (the signal, which should represent a quintet, was not resolved, probably due to the solvent effect [7-8]). Therefore, we investigated the effect of the nature of the solvent, the reagent ratio, and the reaction time on the degree of conversion of the starting compounds (see Table).
|
№ Ex. |
Solvent |
Conversion of C2F5SiMe3*, % (time, h) |
||
|
1 |
Diglyme |
34 (6) |
69 (28) |
74 (72) |
|
2 |
DME |
36 (8) |
56 (32) |
61 (72) |
|
3 |
DME C2F5SiMe3 (3 eq.) |
65 (7) |
81 (31) |
91 (100) |
|
4 |
DME C2F5SiMe3 (5 eq.) |
- |
- |
94 (90) |
|
5 |
THF |
47 (7.5) |
- |
66 (79) |
|
6 |
Et2O |
17 (7) |
- |
24 (79) |
|
7 |
1,4-Dioxane |
0 (6) |
0 (30) |
- |
|
8 |
HMDSO |
- |
0 (24) |
- |
|
9 |
NEt3 |
- |
0 (24) |
- |
|
10 |
TMEDA |
- |
0 (24) |
- |
|
*The conversion degree for C2F5SiMe3 was determined based on a data set of the 1H NMR spectra (ratio of signals at δ=0.80 (C2F5SiMe3) and 0.57 (HSiMe3)) and 19F NMR spectra (ratio of signals at δ=-126 (LiC2F5AlH3) and -132 (C2F5SiMe3)). Also, the amount of unreacted LiAlH4 (ratio of signals at δ=116 (LiC2F5AlH3) and 102 (LiAlH4)) was recorded from the 27Al NMR spectra. |
||||
The above data show that the reaction proceeds equally well in strongly solvating donor ethereal solvents such as diglyme, monoglyme, and THF (runs 1-5). At the same time, the conversion of (pentafluoroethyl)trimethyl silane in diethyl ether for a similar time is significantly lower (run 6). In turn, C2F5SiMe3 does not react with LiAlH4 in 1,4-dioxane, most likely, due to the very low solubility of LiAlH4 (solubility of LiAlH4 in 1,4-dioxane at 25°C is 0.1 g in 100 g [9]). No reaction was also observed using hexamethyldisiloxane, triethylamine, and TMEDA as a solvent, probably, for similar reasons (runs 8-10). In terms of the convenience of isolating complex Li[C2F5AlH3], relatively low boiling solvents such as THF and monoglyme are the most suitable. Thus, we have shown that when carrying out the reaction in monoglyme, to achieve almost complete conversion of LiAlH4, a large (3-5 equiv.) excess of C2F5SiMe3 and an increase in the reaction time to 5 days are required (runs 3-4). A further increase in the reaction time to 9 days leads to the accumulation of side-formed fluorine-containing impurities, the content of which in the isolated product increases from ~ 2% (if the reaction is carried out for 5 days) to 12% (during the reaction time of 9 days). Being carried out under optimal conditions, with the subsequent removal of volatiles in vacuo and toluene purification of the product, the reaction gives a solvated Li[C2F5AlH3]2DME complex. Similarly, Li[C2F5AlH3]2ТHF complex was obtained.
It should be noted that we failed to carry out this reaction using a slight excess of (pentafluoroethyl)trimethyl silane up to complete conversion of LiAlH4 by shifting the equilibrium towards the formation of products through distilling off low-boiling HSiMe3 (bp 7°C). The process almost terminates at 70-80% conversion, and only using a large excess of C2F5SiMe3 (>3 equiv.) is it possible to achieve a high degree of LiAlH4 conversion.
Ethereal solvents (Et2O, THF) can be completely removed from their complexes with LiAlH4 by heating in a dynamic vacuum (~0.5 Torr) at a temperature of 60-70°C [9-10]. However, an attempt to remove a solvent from Li[C2F5AlH3]2DME by performing such a procedure at 55‑60оС leads to decomposition of this complex hydride accompanied by a strong explosion (decomposition point of LiAlH4 >100-150°C [9-10]).
Thus synthesized Li[C2F5AlH3]2DME is a colorless liquid with the following spectral characteristics. Hydride protons appear in the 1H NMR spectra as a broad singlet at 3.5 ppm (for the THF complex, the signal is sharper, with δ=3.2 ppm), which is quite close to the value of a chemical shift of protons in LiAlH4 (δ=2.9 ppm (in DME)) [11]. The 19F NMR spectrum contains two pairs of signals corresponding to the C2F5 moiety (δ: -126 (br.s, 2F, CF2Al), -83.8 (s, 3F, CF3) and δ: -128 (br.s, 2F, CF2Al), -84 (s, 3F, CF3), in a ratio of 2.5:1. Accordingly, there are two signals for CF3 moeties in the 13С{1H} NMR spectrum appearing as a quartet of triplets at ~123 ppm (1JCF = 283 Hz, 2JCF = 30 Hz) and a highly broadened triplet at 132 ppm corresponding to the aluminum-bonded CF2 moiety. The 7Li and 27Al NMR spectra show a singlet of -0.7 ppm and a highly broadened singlet at 117 ppm respectively. When recording the 19F and 27Al NMR spectra in a dilute solution in monoglyme, the form of the spectra becomes similar to that of the reaction mixture. Namely, in the fluorine NMR spectrum, signals for nearly one C2F5 moiety are observed (δ: -126 (br.s, 2F, CF2Al), -84.3 (s, 3F, CF3)) (the content of the second pairs of signals for the C2F5 moiety decreases from 25% to 6%), while the 27Al NMR spectrum contains a highly broadened quartet at 117 ppm (1JAlH = ~170 Hz) (for comparison, the 27Al-1H coupling constant in the AlH4- anion is 170-175 Hz [10]).
When stored in a sealed ampoule for several weeks, the liquid Li[C2F5AlH3]2DME complex transforms into a crystalline-like but in fact a gel-like product. Raman spectra of the liquid complex and the gel-like compound formed 2 weeks later are identical, which suggests that the change in the state of aggregation does not occur through the decomposition of the complex. When refrigerated (~ -25°C), the liquid complex transforms into a white crystalline substance that can be stored for a long time without visible decomposition.
In the presence of chelating agents such as TMEDA or DABCO in ethereal solvents, Li[C2F5AlH3] decomposes, so we failed to obtain complexes of the type LiAlH4-TMEN [11] (TMEN = TMEDA), although triethylamine does not react with Li[C2F5AlH3] under these conditions.
(n-Heptafluoropropyl)trimethyl silane reacts with LiAlH4 similarly. At the same time, when carrying out the reaction of nC3F7SiMe3 with LiAlH4, under optimal conditions (DME, 5 equiv. of the silane, 5 days), a noticeable decrease (to 77%) in the yield of Li[nC3F7AlH3].2DME along with an increase in the yield of fluorine-containing by-products up to 7-12% is observed. The resulting n-heptafluoropropyl complex Li[nC3F7AlH3]2DME is also a liquid and its spectral characteristics are identical to those for the complex bearing C2F5 moiety.
With a further elongation of the perfluoroalkyl substituent, reactions begin to dominate affording fluorine-containing by-products. Thus, nC6F13SiMe3 reacts with LiAlH4 in THF for 2 days to give, according to 1H, 19F, and 27Al NMR spectra, the corresponding (n‑perfluorohexyl)trihydroaluminate complex in ~30% yield. However, as the reaction proceeds further, significant amounts of by-products begin to appear in the reaction mixture, which makes it impossible to isolate the Li[nC6F13AlH3]2THF complex in pure form.
Similar changes in the reactivity of (perfluoroalkyl)trimethyl silanes associated with the length of the perfluoroalkyl radical were also observed earlier. For example, it is known that CF3SiMe3 and C2F5SiMe3 perfluoroalkylate B(OMe)3 almost quantitatively in the presence of KF in THF or diglyme solution to afford corresponding borate salts. At the same time, nC3F7SiMe3 reacts with B(OMe)3 under similar conditions to give K[nC3F7B(OMe)3] in a very low yield [12].
It is known from the literature, that Na[EtAlH3] is unstable in solution and exists as an equilibrium mixture with the products of its disproportionation, namely, NaAlH4 and Na[Et2AlH2], which was established from the 27Al NMR spectrum containing three signals corresponding to these compounds [13]. Unlike its ethyl analog, Na[iBuAlH3] is stable and can be obtained via the reaction between NaAlH4 and NaAliBu4 or AliBu3 [13]. Other examples of lithium alkyl- and aryltrihydroaluminates have also been reported, the stability of which is determined by the steric volume of an aliphatic or aromatic substituent on the aluminum atom, e.g., Li[(Me3Si)3CAlH3] and Li[(Me2PhSi)3CAlH3] [14], and also Li[(2,4,6-iPr3C6H2)AlH3] and Li[(2,4,6-tBuC6H2)AlH3] [15].
We found that Li[C2F5AlH3] and Li[nC3F7AlH3] solvated with DME or THF are fairly stable compounds and can be obtained in pure form from the corresponding (perfluoroalkyl)trimethyl silanes and LiAlH4 in good yields. In this reaction, RFSiMe3 react with LiAlH4 like aliphatic derivatives of alkali, alkaline-earth metals, Zn, Cd, B, and Al [9]. However, we failed to obtain pure forms of lithium (perfluoroalkyl)trihydroaluminates bearing CF3 and nC6F13 moieties, probably due to their instability under reaction conditions. It is interesting to note that recently, two papers have been simultaneously published on the synthesis of the tetrakis(pentafluoroethyl)aluminate anion, [Al(C2F5)4]- [16, 17], which, according to the authors, is the first characterized perfluoroalkyl aluminum derivative containing an RF-Al bond. In this case, one of the synthetic procedures to obtain Li[(C2F5)4Al], a salt which is unstable in ether solution or solid state and solvated with Et2O, comprised the reaction of (C2F5)3SiMe or (C2F5)3SiH with LiAlH4 to produce exclusively a tetrakis-substituted product and corresponding hydrosilanes [16]. In the reaction of C2F5SiMe3 with LiAlH4 discovered by us, the formation of poly-C2F5-substituted aluminates was not observed.
To conclude, a general possibility of synthesizing stable perfluoroalkyl aluminum derivatives by replacing the substituent in the coordination sphere of the tetrahedral aluminum anion with a perfluoroalkyl group has been demonstrated for the first time, which opens up prospects for further study of the properties of this, as yet exotic, class of compounds. Even though the first, the most natural idea of using Li[C2F5AlH3]2DME for the synthesis of lithium (pentafluoroethyl) alkoxyaluminates has not been fully realized by us, since reactions with alcohols lead to partial substitution of the C2F5 group furnishing the difficultly separable mixture of products, one can expect that the replacement the Li cation with bulky organic cations such as PNP or EtP4H (as in [16]) can be a possible approach to obtain [C2F5Al(OR)3]- anions.
Experimental
1H, 13C, 19F, 27Al, 29Si, and 7Li NMR spectra were recorded on a Bruker AM-300, Bruker AVANCE-400, Bruker AVANCE-500, or Bruker AVANCE-600 spectrometers with 300.13, 400.13, 500.13, and 600.22 MHz working frequencies for 1H, respectively. The frequencies of other nuclei, depending on the instrument used, are given in the Experimental Section below when referring to the NMR spectra of the corresponding compounds. Chemical shifts were referenced to the residual proton chloroform peak (7.26 ppm in CDCl3) and reported in ppm units relative to TMS. Chemical shifts in 13C NMR spectra were referenced to the 13С nucleus peak (77.0 ppm in CDCl3) and reported in ppm units relative to TMS. Chemical shifts in 19F NMR spectra were reported in ppm units relative to an external CFCl3 reference. Chemical shifts in 27Al NMR spectra were reported in ppm units relative to an external Al(NO3)3 reference. Chemical shifts in 29Si NMR spectra were reported in ppm units relative to an external TMS reference. Chemical shifts in 7Li NMR spectra were reported in ppm units relative to an external LiCl reference. Positive values of chemical shift correspond to the downfield shift of the indicator nucleus signal. Raman spectra were recorded on a Jobin Yvon LabRam spectrometer. Elemental analysis was performed in the Laboratory of Microanalysis of INEOS RAS.
All experiments were carried out in an inert atmosphere using anhydrous solvents and standard working procedures. Before use, commercially available LiAlH4 was purified by the standard method [9]: it was dissolved in Et2O, the insoluble grey residue was allowed to settle down, the clear solution was separated, evaporated in vacuo to dryness or to a concentrated solution from which the hydride was precipitated by adding toluene and separated by filtration. The resulting white powdered LiAlH4 was dried at 60-65°C (<0.5 Torr). (Perfluoroalkyl)trimethylsilanes: CF3SiMe3, C2F5SiMe3, nC3F7SiMe3, and nC6F13SiMe3 are prepared by the procedure developed by us earlier [18-19].
To analyze the reaction mixtures, ~0.3 mL of the reaction solution was sealed in a glass ampoule (~3 mm diameter and 110-130 mm length). This ampoule was then placed in a standard 5 mm NMR ampoule, CDCl3 was added as an external standard, and the reaction mixture was analyzed by NMR.
Synthesis of Li[C2F5AlH3]2DME
To a stirred suspension of LiAlH4 (1 g, 0.026 mol) in DME (50 mL), C2F5SiMe3 (25 g, 0.13 mol) is added. The resulting mixture is stirred for several hours until homogeneous, and then kept for 5 days, periodically stirring the solution. Volatiles are then removed in vacuo (<0.5 Torr) and collected in a trap to provide a white solid, which turns into a liquid upon prolonged drying in vacuo (<0.5 Torr).
To the residue, toluene (20 mL) is added and the resulting mixture is stirred until dissolution and the formation of a liquid two-phase mixture, from which the solvent is distilled off in vacuo (<0.5 Torr). The liquid residue is dried for several hours in vacuo (<0.5 Torr) to give a colorless liquid (7.8 g, yield 90%).
Found, %: C, 35.52; H, 6.73. C10H23AlF5LiO4. Calculated, %: C, 35.73; H, 6.90.
1H NMR (500.13 MHz) δ: 3.45-3.65 (br.s, 3H, AlH3), 3.95 (s, 12H, OCH3), 4.14 (s, 8H, CH2CH2);
19F NMR (470.59 MHz) δ: two sets of signals at -128 (s, 2F, CF2), -84 (s, 3F, CF3), and -126 (br.s, 2F, CF2), -83.8 (s, 3F, CF3) in a ratio of 1:2.5;
13С{1H} NMR (125.75 MHz) δ: 59 (s, CH3O), 70 (s, CH2O), 123 (qt, 1JCF = 283 Hz, 2JCF = 30 Hz, CF3), 132 (br.t, CF2Al);
27Al NMR (104.26 MHz) δ: 117 (br.s);
7Li NMR (155.5 MHz) δ: -0.66 (s).
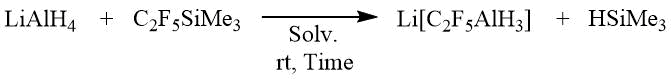
Raman spectrum of Li[nC3F7AlH3]2DME
Synthesis of Li[nC3F7AlH3]2DME.
Synthesized similarly to Li[nC3F7AlH3]2DME. Colorless viscous liquid, which is partially solidified at -25°C.
Found, %: C, 34.21; H, 6.26. C11H23AlF7LiO4. Calculated, %: C, 34.21; H, 6.00.
1H NMR (500.13 MHz) δ: 3.45-3.65 (br.s, 3H, AlH3), 3.9 (s, 12H, OCH3), 4.1 (s, 8H, CH2CH2);
19F NMR (282.4 MHz) δ: two pairs of signals in the CF2 region -128 (s, 2F, CF2CF2Al), -126.3 (s, 2F, CF2CF2Al), and -126.6 (s, 2F, CF2CF2Al), -126.1 (s, 2F, CF2CF2Al) in a ratio of 1:4, -81.3 (s, 3F, CF3);
13С{1H} NMR (125.75 MHz) δ: 59 (s, CH3O), 70 (s, CH2O), 112 (tq, 1JCF = 251 Hz, 2JCF = 28 Hz, CF2), 119 (qt, 1JCF = 288 Hz, 2JCF = 38 Hz, CF3), 135 (br.t, CF2Al);
27Al NMR (104.26 MHz) δ: -117 (br.s);
7Li NMR (155.5 MHz) δ: -0.64 (s).
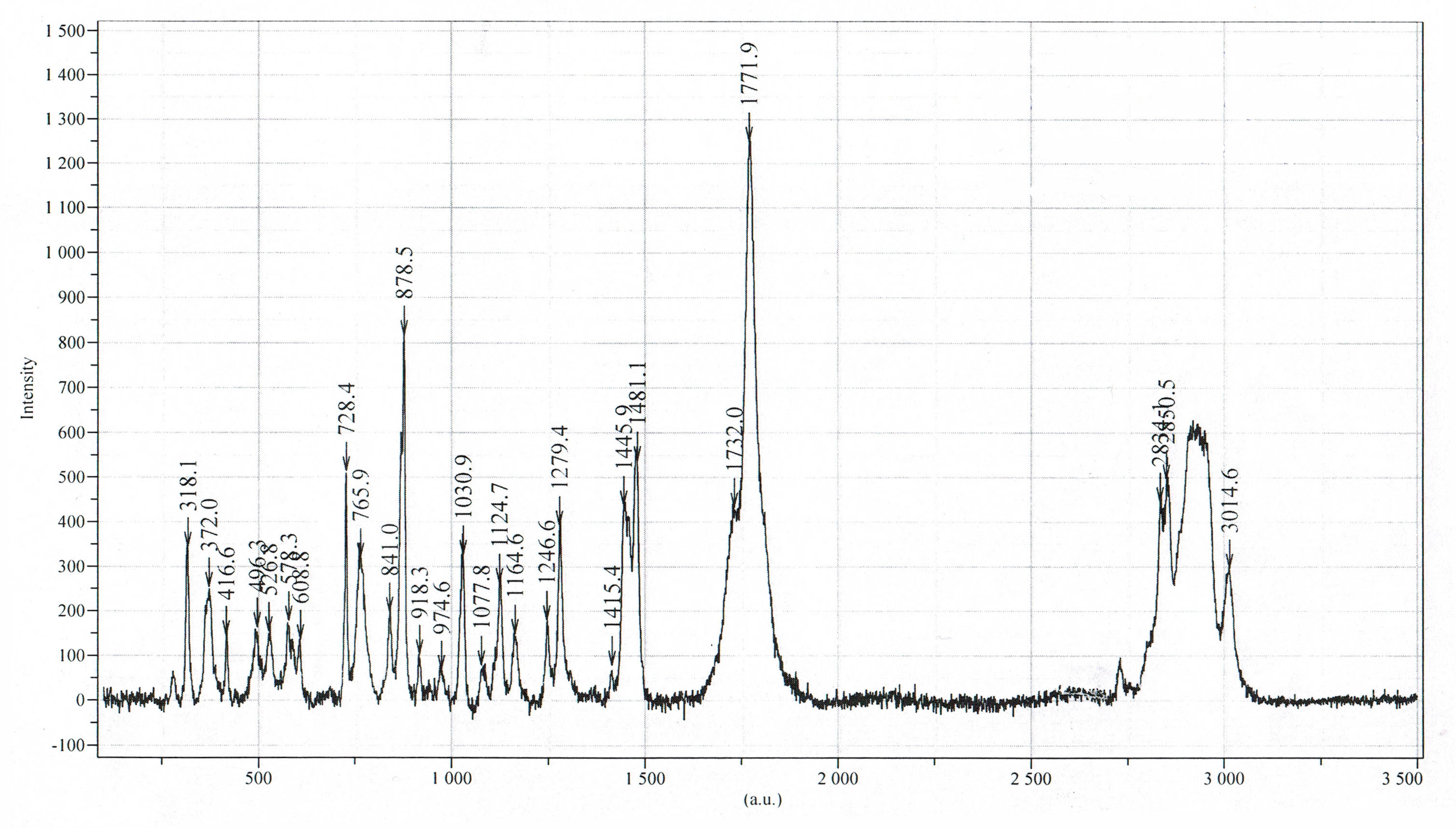
Raman spectrum of Li[nC3F7AlH3]2DME
Acknowledgements
This study was financially supported by the Ministry of Science and Higher Education of the Russian Federation. The contribution of the Center for Molecule Composition Studies of INEOS RAS is gratefully acknowledged.
References
- A.A. Tyutyunov, V.E. Boyko, S.M. Igoumnov, Fluorine Notes, 2011, 1(74).
- P.T. Hennig, J.A.P. Sprenger, L.N. Schneider, N.V. Ignat’ev, M. Finze, Chem. Commun., 2019, 55, 6110-6113.
- The results of the study were presented at the 57th traditional contest-conference of research works of INEOS RAS, 2012.
- J. Pinkas, H.W. Roesky, J.Fluor.Chem., 2003, 122, 125-150.
- H.W. Roesky, A. Stasch, H. Hatop, C. Rennekamp, D.H. Hamilton, M. Noltemeyer, H.-G. Schmidt, Angew. Chem. In. Ed., 2000, 39, 171-173.
- A.G. Avent, W.-Y. Chen, C. Eaborn, I.B. Gorrell, P.B. Hitchcock, J.D. Smith, Organometallics, 1996, 15, 4343-4345.
- H. Noth, R. Rurlander, P. Wolfgardt, Zeitschrift fur Naturforschung B, 1980, 35, 31-41.
- H. Noth, Zeitschrift fur Naturforschung B, 1980, 35, 119-124.
- E.C. Ashby, Adv. Inorg. Chem. Radiochem., 1966, 8, 283-335.
- V.P. Tarasov, G.A. Kirakosyan, Russ. J. Inorg. Chem., 2008, 53, 2048-2081.
- M.M. Andrianarison, A.G. Avent, M.C. Ellerby, I.B. Gorrell, P.B. Hitchcock, J.D. Smith, D.R. Stanley, J. Chem. Soc., Dalton Trans., 1998, 249-254.
- N.Yu. Adonin, V.V. Bardin, H.-J. Frohn, Z. Anorg. Allg. Chem., 2007, 633, 647-652.
- V.V. Gavrilenko, M.I. Vinnikova, V.A. Antonovich, L.I. Zakharkin, Bull. Acad. Sci. USSR, Div. Chem. Sci., 1982, 31, 2084-2087.
- C. Eaborn, I.B. Gorrell, P.B. Hitchcock, J.D. Smith, K. Tavakkoli, Organometallics, 1994, 13, 4143-4144.
- R.J. Wehmschulte, J.J. Ellison, K. Ruhlandt-Senge, P.P. Power, Inorg.Chem., 1994, 33, 6300‑6306.
- N. Tiessen, B. Neumann, H.-G. Stammler, B. Hoge, Chem. Eur. J., 2020, 26, 13611-13614.
- L.A. Bischoff, J. Riefer, R. Wirthensohn, T. Bischof, R. Bertermann, N.V. Ignat’ev, M. Finze, Chem. Eur. J., 2020, 26, 13615-13620.
- S.M. Igoumnov, V.K. Men’shikov, V.E. Boyko, A.A. Tyutyunov, S.R. Sterlin, Fluorine Notes, 2012, 6(85).
- V.E. Boyko, A.A. Tyutyunov, V.L. Don, S.M. Igoumnov, Fluorine Notes, 2013, 6(91).
ARTICLE INFO
Received 22 October 2021
Accepted 29 October 2021
Available online December 2021
Recommended for publication by PhD M. A. Manaenkova
Fluorine Notes, 2021, 139, 1-2
