Received: July 2022
DOI 10.17677/fn20714807.2022.04.02
Fluorine Notes, 2022, 143, 3-4
SYNTHESIS OF 2-OR 4- FLUORINE-SUBSTITUTED ARYLAMIDES
T.P. Vasilyeva*, V.I. Dyachenko, D.V. Vorobyeva
A.N. Nesmeyanov Institute of Organoelement Compounds RAS,
Russian Federation, 119334, Moscow, Vavilov str. 28/1.
Fax: +7(499) 135 5085. E-mail: d-20@mail.ru
Abstract: The reactions of monofluorosubstituted benzoyl chlorides with cyclic, heterocyclic and aromatic amines in various conditions were studied. It has been shown that higher yields of aminolysis products are achieved at an equimolar ratio of reagents in pyridine medium (at 20C) or in the presence of catalytic amounts of pyridine on heating. As a result, preparative methods for the synthesis of 2- or 4-fluorosubstituted aryl amides suitable for scaling up have been developed.
Keywords: amines, 2-fluorobenzoyl chloride, 4-fluorobenzoyl chloride, aminolysis, fluorinated benzoyl amides, pyridine.
Previously, we developed the method for preparation of carboxylic acids esters containing aryl and trifluoromethyl groups in the -position [1]. In continuation of these studies, we studied approaches to synthesis of arylamides containing one fluorine atom in the aromatic nucleus. It is known that fluoroaryl-containing drugs have increased activity compared to hydrogenated analogues [2]. At the same time, the amide bond is the most important functional group that is part of natural substances (proteins), pharmaceuticals and polymers [3]. According to latest data, at least half of all routes for synthesis of bioactive substances in medicinal chemistry contain at least one stage of amide bond formation [4].
Several methods for preparation of monofluorosubstituted arylamides have been described. Thus, 4-fluorobenzoic acid phenylethylamide 1 was synthesized by oxidative amidation of 4-fluorobenzyl alcohol [5] or 4-fluorobenzaldehyde [6] with phenylethylamine in the presence of 2-3 mol. % of a complex tungsten-containing catalyst and tert-butyl hydroperoxide at 90°C. In [7], amide 1 was obtained by action of phenylethylamine on 4-fluorobenzoyl chloride, but synthesis procedure and spectra of 1 are not shown. Phenylethylamide 1 is the starting compound in synthesis of biologically active 1-(4-fluorophenyl)-1,2,3,4-tetrahydroisoquinolines [7, 8] used, in particular, in gynecology [8].
Among the known monofluorosubstituted arylamides, the most popular is 4-fluorobenzoic acid morpholide 2, which turned out to be an active acylating agent and useful building block for preparation of both aldehydes and ketones from various derivatives of carboxylic acids [9, 10] and fluorine-containing mono- and bis(porphyrins) [11], pyrrole-substituted fungicides [12], arylboronic acids and esters. The latter compounds are used in synthesis of biologically active substances [13].
Morpholide 2 was previously obtained by aminocarboxylation of 4-fluorobromobenzene with morpholine in the presence of K2CO3 and two catalysts—Pd nanoparticles at the zeolite–imidazole support and bis(2-diphenylphosphine)ether [14] (see Scheme 1).
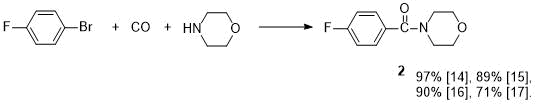
Scheme 1. Yields of morpholide 2 with various catalysts.
In [15], the compound 2 was also synthesized according to Scheme 1, but Pd nanoparticles at another support (copolymer of melamine and formaldehyde) were used as a catalyst, and this reaction was carried out in the presence of K3PO4. There are reports about synthesis of 2 (according to Scheme 1) under microwave irradiation using Hunig's base (DIPEA) in the presence of catalytic amounts of Pd(PPh3)2Cl2 [16] or in the presence of iridium complex [17]. When using 4‑fluoroiodobenzene (instead of bromide) as the starting compound under UV irradiation, amide 2 is formed according to Scheme 1 in 60% yield in the absence of a catalyst and a base [18]. The preparation of morpholide 2 by catalytic amidation (by means morpholine) of 4-fluorobenzaldehyde in the presence of rhodium catalyst and oxidizing agent [19], or using samarium complexes [20, 21], as well as through intermediate acylhydrazides [22], was reported. Direct synthesis of morpholide 2 in yield of 86-88% by coupling reaction of p-fluorobenzoic acid with morpholine in the presence of phenylsilane [3] or mesoporous silica gel [23] has been described. It is possible to obtain 2 by preliminary acylation of this acid with N,N-carbonyldiimidazole to give acylimidazole, which was further treated with morpholine [24]. A mixture of ethyl 4-fluorobenzoate and morpholine placed in a steel ball mill gave the amide 2 in the presence of But-OK in yield 55% [4].
The best yield of 2 (92%) was obtained by reaction of 4-FC6H4COOEt ester with reaction product of morpholine and diisobutylaluminum hydride [9]. Transamidation of secondary amide FC6H4CON(Ph)Boc under the action of morpholine synthesized morpholide 2 in yield 89% [25]. There are reports about preparation of 4-fluorobenzoyl chloride by 2 aminolysis [11-13]. We improved the methods of synthesis and isolation of the product, as a result, we managed to increase the yield from 56 to 88% and also the purity of morpholide 2.
Cyclohexylamide of 4-fluorobenzoic acid was previously obtained by oxidative amidation of 1-(p-fluorophenyl)-2-azidoethanol with cyclohexylamine in the presence of Ph-I(OAc)2 (in yield 80%) [26], as well as by radical amidation of cyclohexane with 4-fluorobenzamide in the presence of di-tert-butyl peroxide and the catalyst - copper acetylacetonate (in yield 92%) [27] or in the presence of mixture of CuI and dimethoxyphenanthroline (in yield 78%) [28]. On reaction of 4-FC6H4CONH2 with iodocyclohexane in the presence of lithium tert-butoxide and the catalyst CuI under UV irradiation, the yield of cyclohexylamide was 91% [29].
In contrast to 4-fluorobenzoylamides, in the literature there are few reports about synthesis of 2-fluoro-substituted analogs. For example, it has recently been shown that the Pd-catalyzed reaction of N-benzoylpiperidine with N-fluorobenzenesulfonylimide in the presence of nitrate anion gives 2-fluorobenzoylpiperidine [30] (see Scheme 2).
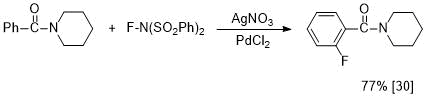
Scheme 2. С,Н-fluorination of N-benzoylpiperidine.
The latter compound is a synthon for electrochemical preparation of α-alkoxy- or α,α-dialkoxy derivatives of piperidine [31]. Some o-fluorinated benzamides are known to be anticancer drugs [30]. The aim of this paper was the synthesis of fluorosubstituted arylamides, which may be of interest for medicinal chemistry. Although many new methods have been found to date, the reaction of acyl chlorides with amines remains one of the most efficient and widely used methods for synthesis of amides. We have developed the preparation of amides of 4-fluorobenzoic acid - phenylethylamide 1, morpholide 2, furfurylamide 3, hexamethyleneimide 4, dicyclohexylamide 5, as well as amides of 2-fluorobenzoic acid - furfurylamide 6 and piperidide 7 by reaction of corresponding acid chlorides with amines at a reagent ratio of 1:2 (Method A, see Scheme 3).
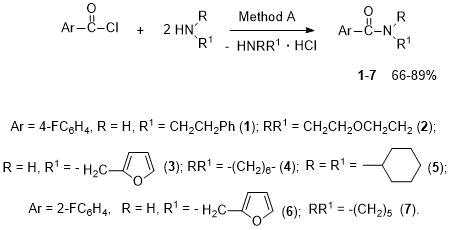
Scheme 3. Synthesis of arylamides 1-7 (Method A).
In addition, these arylamides were synthesized from ArCOCl and amines at an equimolar ratio of reagents. Using reactions with cyclic amines (cyclohexylamine, hexamethyleneimine) as an example, it was shown that, in the presence of excess of pyridine, the corresponding amides 8, 9 are formed in higher yields than by Method A. The products were isolated by acidifying the reaction mixture and extracting with ethyl acetate (Method B, see Scheme 4).
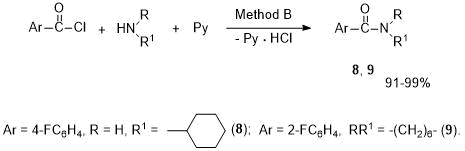
Scheme 4. Synthesis of arylamides 8, 9 in pyridine medium (Method B).
We have studied the possibility of carrying out this reaction without pyridine excess. Thus, the aminolysis of 4-fluorobenzoyl chloride with equimolar amount of o-phenetidine produces a mixture (1:1) of amide 10 and amine hydrochloride, however, after refluxing in benzene in the presence of catalytic amounts of pyridine, product 10 was obtained in high yield (Method C, see Scheme 5). The disadvantage of this method is process duration (7 h).
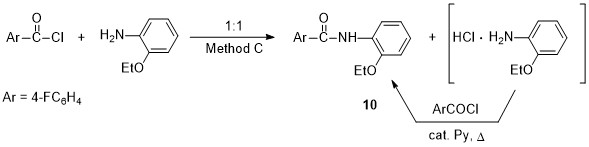
Scheme 5. Py-catalyzed reaction of equimolar amounts of reagents (Method C)
In most cases, the purification of obtained compounds was achieved by passing a solution in organic solvent through a small layer of silica gel. Synthesized by us amides 3-6 and 9, 10 are a new compounds, the composition and structure of which were proved by 1H and 19F NMR spectroscopy data and elemental analysis. In 19F NMR spectra of 4-fluorine-containing compounds 3-5 and 10, the fluorine signal appears in the region of -107.8 -112.2 ppm, and for 2-fluoro-substituted amides 6, 7, 9, a singlet at -1150-115.4 ppm is characteristic.
Thus, in this paper, the preparative methods for obtaining of fluorosubstituted arylamides (both new and previously obtained by other methods) have been developed. Since the organofluorine compounds are used in various areas of medicine and agrochemistry [32], and many drugs has an amide function [4, 21, 30], the synthesis of new fluorine-containing amides is the relevant objective.
Experimental part
NMR spectra were recorded via Bruker Avance-200, Bruker Avance-300 and Bruker Avance-400 spectrometers at operating frequencies of 200, 300 and 400 MHz and internal standard SiMe4 for 1H NMR, and at operating frequency of 282 MHz for 19F NMR and internal standard CFCl3). Elemental analysis was performed in the Laboratory of Microanalysis of INEOS RAS. The reaction progress was monitored by TLC test on Merck plates (silica gel 60 F254, 0.25 mm). Used 2- or 4-fluorobenzoyl chlorides are commercially available reagents. All used solvents were purified according to standard procedures. The RF values of obtained compounds were determined in the system «acetone-СCl4» (at a ratio 1:3).
Aminolysis of fluorobenzoyl chlorides. Method A. Synthesis of arylamides 1-7.
To a solution of 16 mmol of amine in 19 ml of benzene (or diethyl ether) was added 7.6 mmol of ArCOCl in 8 ml of benzene (or diethyl ether) while cooling with cold water and with stirring. The next day, the formed precipitate was removed by filtration, the mother solution was washed with water and dried over MgSO4. After removal of MgSO4, this solution was filtered again through a small layer of silica gel (3 mm). After evaporation of solvents in vacuo, the pure products 1-7 were obtained. To isolate of low-melting amides 2, 4 and 7, the residue was additionally treated with cold hexane.
4-Fluoro-N-phenethylbenzamide (1).
Yield 73%, white crystals, m. p. 127С. 1Н and 19F NMR spectra in CDCl3 coincide with described in literature [5, 33].
4-(Fluorophenyl)(morpholino)methanone (2).
Yield 88%, low melting white crystals, RF = 0.34 (solution in diethyl ether). 1Н and 19F NMR spectra in CDCl3 coincide with described in literature [3, 4, 11, 25].
Fluoro-N-(furan-2-ylmethyl)benzamide (3).
Yield 81%, beige crystals, m.p. 120-122С. 1H NMR (200 MHz, CD3OD,δ, ppm, J/Hz): 4.45 (s, 2H, CH2); 4.81 (s, 1H, NH); 6.25 (d, 2H, furan, J=6.0); 7.09 (t, 2H, Ar, J = 9.0); 7.34 (s, 1H, furan); 7.75-7.90 (m, 2H, Ar). 19F NMR (282 MHz, CDCl3, δ, ppm): -107.82 (s, 1F, F-Ar). Founded (%): C, 65.56; H, 4.44; N, 6.23. C12H10FNO2. Calculated (%): C, 65.75; H, 4.57; N, 6.39.
Azepan-1-yl (4-fluorophenyl) methanone (4).
Yield 69%, transparent crystals, m.p. 30С, RF = 0.48 (solution in diethyl ether). 1H NMR (400 MHz, CDCl3, δ, ppm, J/Hz): 1.50-1.94 (m, 8H, 4CH2); 3.39 (br.s, 2H, CH2N); 3.63 (br.s, 2H, CH2N); 7.08 (t, 2H, Ar, J = 10.0); 7.37-7.41 (m, 2H, Ar). 19F NMR (282 MHz, CDCl3, δ, ppm): -111.29 (s, 1F, F-Ar). Founded (%): C, 70.36; H, 7.13; N, 6.09. C13H16FNO. Calculated (%): C, 70.59; H, 7.24; N, 6.33.
N,N-Dicyclohexyl-4-fluorobenzamide (5).
Yield 66%, white crystals, m.p. 113-116С, RF = 0.78 (solution in methanol). 1H NMR (300 MHz, CDCl3, δ, ppm, J/Hz): 0.83-2.48 (m, 20H, 10CH2); 3.12 (br.s, 2H, 2CH); 7.00 (t, 2H, Ar, J = 9.0); 7.22 (t, 2H, Ar, J = 3.0). 19F NMR (282 MHz, CDCl3, δ, ppm): -112.18 (s, 1F, F-Ar). Founded (%): C, 75.53; H, 8.82; N, 4.54. C19H26FNO. Calculated (%): C, 75.25; H, 8.58; N, 4.62.
2-Fluoro-N-(furan-2-ylmethyl)benzamide (6).
Yield 89%, yellow crystals, m.p. 42-44С. 1H NMR (200 MHz, CD3OD, δ, ppm, J/Hz): 4.50 (s, 2H, CH2N); 4.84 (s, 1H, NH); 6.19-6.33 (m, 2H, furan); 7.06-7.26 (m, 2H, Ar); 7.36-7.53 (m, 2H, Ar); 7.61-7.73 (m, 1H, furan). 19F NMR (282 MHz, CDCl3, δ, ppm): -115.0 (s, 1F, F-Ar).
Founded (%): C, 65.54; H, 4.42; N, 6.15. C12H10FNO2. Calculated (%): C, 65.75; H, 4.57; N, 6.39.
2-Fluorophenyl(piperidin-1-yl)methanone (7).
Yield 83%, low melting white crystals, RF = 0.60 (solution in benzene). 1Н and 19F NMR spectra in CDCl3 coincides with described in literature [30].
Method B. Synthesis of arylamides 8, 9.
To a solution of 7 mmol of amine and 3.4 ml (42 mmol) of pyridine in 15 ml of benzene, 7 mmol of ArCOCl in 10 ml of benzene was added with stirring under cold water. The next day, the reaction mixture was treated with dilute 4% hydrochloric acid to pH1 (20 ml). The benzene solution was separated, the aqueous layer was extracted twice with ethyl acetate. The combined organic layers were dried over MgSO4 and filtered through a small layer of silica gel (2 mm). The filtrate then was evaporated to dryness in vacuo to give amides 8 and 9.
N-cyclohexyl-4-fluorobenzamide (8).
Yield 99%, light yellow crystals, m.p. 140С. 1H NMR spectrum in CDCl3 coincides with described in literature [26-29]. 19F NMR (282 MHz, CDCl3, δ, ppm): -108.62 (s, 1F, F-Ar).
Azepan-1-yl(2-fluorophenyl)methanone (9).
Yield 91%, light yellow oil, RF = 0.55 (solution in benzene). 1H NMR (300 MHz, CDCl3, δ, ppm): 1.25-1.81 (m, 8H, 4CH2); 3.30 (br. s, 2H, CH2N); 3.67 (br. s, 2H, CH2N); 7.06-7.32 (m, 4H, Ar). 19F NMR (282 MHz, CDCl3 δ, ppm): -115.41 (s, 1F, F-Ar). Founded (%): C, 70.46; H, 7.16; N, 6.15. C13H16FNO. Calculated (%): C, 70.59; H, 7.24; N, 6.33.
Method C. Synthesis of N-(2-ethoxyphenyl)-4-fluorobenzamide (10).
To a solution of 1.5 g (9.5 mmol) of 4-FC6H4COCl in 10 ml of benzene, 1.36 g (9.9 mmol) of o-fenethidine in 5 ml of benzene was added with cooling by cold water, a precipitate formed. The next day, three drops of pyridine were added to reaction mixture, then it was boiled at 90°C until HCl evolution ceased (7 h). Oil-like residue of unreacted hydrochloride was removed by filtration, the mother solution was passed through a layer of silica gel ( 6 mm). After evaporation of solvent in vacuo, the oil-like residue was treated with hexane. Yield 1.97 g (80%) of amide 10 in the form of pale pink crystals with m.p. 65-67°C, RF = 0.67 (solution in methanol). 1H NMR (300 MHz, CDCl3, δ, ppm, J/Hz): 1.49 (t, 3H, CH3, J = 6.0); 4.16 (q, 2H, CH2, J = 6.0); 6.91 (d, 1H, NH, J = 6.0); 6.98-7.10 (m, 2H, Ar); 7.18 (t, 2H, Ar, J = 9.0); 7.90 (q, 2H, Ar, J = 3.0); 8.52 (t, 2H, Ar, J = 10.5). 19F NMR (282 MHz, CDCl3, δ, ppm): -107.78 (s, 1F, F-Ar). Founded (%): C, 69.26; H, 5.28; N, 5.46. C15H14FNO2. Calculated (%): C, 69.50; H, 5.41; N, 5.41.
Acknowledgments
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (Contract/agreement No. 075-00697-22-03) and was performed employing the equipment of Center for molecular composition studies of INEOS RAS.
References
- Vasilyeva T.P., Vorobyeva D.V., Russ. Chem. Bull. (Int. Ed.), 2018, 67, 1426.
- Ricci G., Ruzziconi R., J. Org. Chem., 2005, 70, 611.
- Morisset E., Chardon A., Rouden J., Blanchet J., Eur. J. Org. Chem., 2020, 388.
- Nicholson W., Barreteau F., Leitch J., Payne R., Priestley I., Godineau E., Battilocchio C., Browne D., Angew. Chem. Int. Ed., 2021, 60, 21868.
- Fu R., Yang Y., Feng W., Ge Q., Feng Y., Zeng X., Chai W., Yi J., Yuan R., Tetrahedron, 2016, 72, 8319.
- Fu R., Yang Y., Zhang J., Shao J., Xia X., Ma Y., Yuan R., Org. Biomol. Chem., 2016, 14, 1784.
- Gray N., Cheng B., Mick S., Lair C., Contreras P., J. Med. Chem., 1989, 32, 1242.
- Paul R., Coppola J., Cohen E., J. Med. Chem., 1972, 15, 720.
- Jeon A., Kim M., Park J., Shin W., An D., Tetrahedron, 2014, 70(29), 4420.
- Park J., Shin W., An D., Bull. Korean Chem. Soc., 2013, 34, 1592.
- Melomedov J., Leupoldt A., Meister M., Laquai F., Heinze K., Dalton Transaction, 2013, 42(26), 9727.
- Massa S., Ragno R., Porretta G., Mai A., Retico A., Artico M., Simonetty N., Archiv der Pharmazie, 1993, 326, 539.
- Cederbalk A., Lysen M., Kehler J., Kristensen J., Tetrahedron, 2017, 73(12), 1576.
- Dang T., Zhu Y., Ngiam J., Ghosh S., Chen A., Seayad A., ACS Catalysis, 2013, 3, 1406.
- Molla R., Iqubal M., Ghosh K., Roy A., Kamaluddin, Islam S., RSC Advances, 2014, 4(89), 48177.
- Cardullo F., Donati D., Merlo G., Paio A., Petricci E., Taddei M., Synlett, 2009, 1, 47.
- Forni J., Micic N., Connell T., Weragoda G., Polyzos A., Angew. Chem., Int., Ed., 2020, 59, 18646.
- Kawamoto T., Sato A., Ryu I., Chem. Eur. J., 2015, 21, 14764.
- Tillack A., Rudloff I., Beller M., Eur. J. Org. Chem., 2001, 523.
- Wang C., Huang L., Lu M., Zhao B., Wang Y., Zhang Y., Shen Q., RSC Advances, 2015, 5(115), 94768.
- Li J., Xu F., Zhang Y., Shen Q., J. Org. Chem., 2009, 74, 2575.
- Maruani A., Lee M., Watkins G., Akhbar A., Baggs H., Shamsabadi A., Richards D., Chudasama V., RSC Advances, 2016, 6(4), 3372.
- Tamura M., Murase D., Komura K., Synthesis, 2015, 47, 769.
- Verma S., Ghorpade R., Pratar A., Kaushik M., Tetrah. Lett., 2012, 53, 2373.
- Rahman M., Li G., Szostok M., J. Org. Chem., 2019, 84, 12091.
- Yu P., Wang Y., Zeng Z., Chen Y., J. Org. Chem., 2019, 84, 14883.
- Teng F., Sun S., Jiang Y., Yu J., Cheng J., J. Chem. Soc., Chem. Commun., 2015, 51(27), 5902.
- Tran B., Li B., Driess M., Hartwig J., J. Am. Chem. Soc., 2014, 136, 2555.
- Do H., Bachman S., Bissember A., Peters J., Fu G., J. Am. Chem. Soc., 2014, 136, 2162.
- Ning X., Lou S., Mao Y., Xu Z., Xu D., Org. Lett. 2018, 20, 2445.
- Mitzlaff M., Warning K., Jensen H., Liebigs J., Ann. Chem., 1978, 1713.
- Kirsch P., Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, 2nd ed., Wiley-VCH, Weinheim, 2013.
- Spratt M., Dorn H., Anal. Chem., 1984, 56, 2038.
ARTICLE INFO
Received 14 July 2022
Accepted 21 July 2022
Available online August 2022
Recommended for publication by PhD M. Manaenkova
eLIBRARY Document Number (EDN) TEOAHM

Fluorine Notes, 2022, 143, 3-4
