Received: July 2022
DOI 10.17677/fn20714807.2022.04.03
Fluorine Notes, 2022, 143, 5-6
A NEW METHOD FOR PREPARING OF 1,2,2-TRIFLUOROSTYRENE
A.V. Sinkoa, S.M. Igumnovab
aA.N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences,
28 Vavilova St., 119991 Moscow, Russian Federation
bP@M-Invest Scientific Production Association, 119991, Leninsky ave. 47, Moscow, Russian Federation.
e-mail: sinkoav@gmail.com
Abstract: A new preparative method for synthesis of 1,2,2-trifluorostyrene by decarboxylation of potassium α-phenyltetrafluoropropionate has been developed.
Keywords: 1,2,2-trifluorostyrene, α-phenyltetrafluoropropionate
1,2,2-Trifluorostyrenes are attractive monomers for preparation of polymers with a fully fluorinated main chain, which gives such polymers a high chemo- and thermal stability. At the same time, the presence of phenyl fragments makes it possible to introduce various functional groups for obtain a wide variety of modified materials. Such modified polymers based on α,β,β-trifluorovinylstyrenes are used, in particular, as materials for membranes used in fuel cells [1-2], dialysis membranes [3], ion-exchange membranes [4], chromatography carriers, films for optical compensation in LCD and OLED displays [5], coatings in lithium-ion batteries [6] and other areas [7].
1,2,2-Trifluorostyrene was first obtained by Cohen in 1949 by a multi-step synthesis involving the acylation of benzene with trihaloacetic acid chloride, catalyzed by Lewis acids, followed by replacement of oxygen by halogens, fluorination, then elimination of halogens or hydrogen halide, leading to formation of a double bond with a total with a yield of about 48% [8].
1,2,2-Trifluorostyrene was also obtained by reaction of aryllithium compounds with tetrafluoroethylene [9]. Cross-coupling of aryl iodides with trifluorovinylzinc or tin, catalyzed by palladium complexes, was proposed; this method also makes it possible to obtain trifluorostyrenes containing various functional groups in the ring [10-15]. However, in addition to the complexity of operations with palladium complexes, as well as their high cost, the starting trifluorovinyl iodide or bromide necessary for preparation of trifluorovinyl organometallic reagents, are compounds that are difficult to obtain.
Also previously are described the interaction reactions (catalyzed by various palladium complexes) of tetrafluoroethylene with zinc aryl derivatives [15], with aryl iodides with aryl Grignard reagent [16], with trimethoxyphenylsilane [17], or with derivatives of phenylboronic acid [18].
Becker in 1979 discovered the reaction of obtaining α-oxides by dehydrochlorination of tertiary polyfluorochlorocarbinols received by interaction of polyfluorochloroacetones with benzene according to Friedel-Crafts reaction, the opening of such an oxide provided a rather convenient way to styrene. Isomerization of oxide (1) under the action of bases gave the corresponding fluoroanhydride (2), then salt (3) and by its pyrolysis - gave 1,2,2-trifluorostyrene (4) [19].
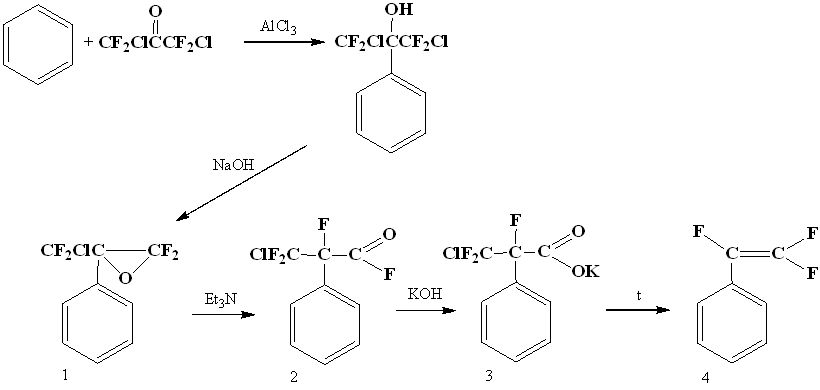
A two-stage method to get of trifluorostyrene is also known, including thermal addition of 1,2-dichloro-2-iodo-1,1,2-trifluoroethane to benzene in an autoclave at 200°C and subsequent dechlorination of resulting trifluorodichloroethylbenzene (5) with zinc [20].
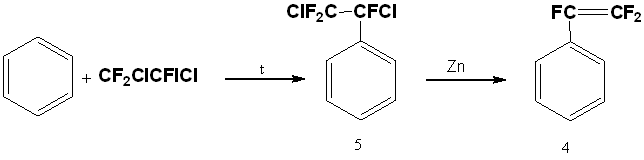
All described synthesis methods of trifluorostyrene (4) are multistage or require the use of special equipment or expensive reagents and work in an inert atmosphere, so the search for new methods for its synthesis is an urgent task.
Initially, we wanted to modify the method [20] so that it would be possible to implement the circuit without using an autoclave. To obtain (5), we tried to combine 1,2-dichloro-2-iodo-1,1,2-trifluoroethane and iodobenzene at metallic copper in DMF.
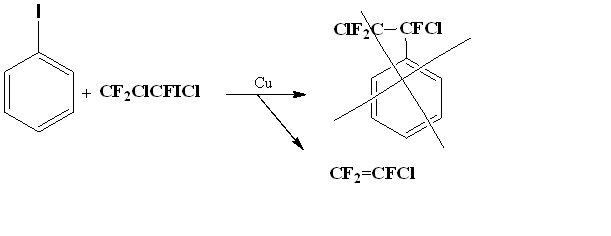
However, we did not observe the formation of an addition product, but only deiodchlorination of 1,2-dichloro-2-iodo-1,1,2-trifluoroethane with formation of trifluorochloroethylene.
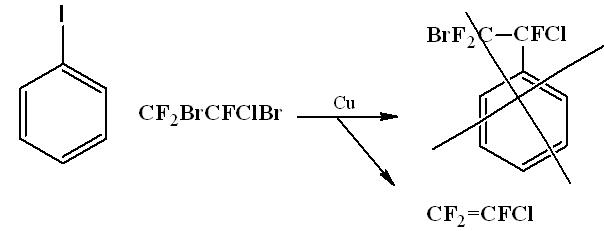
The same situation happened when trying to react with iodobenzene 1,2-dibromo-2-chloro-1,1,2-trifluoroethane.
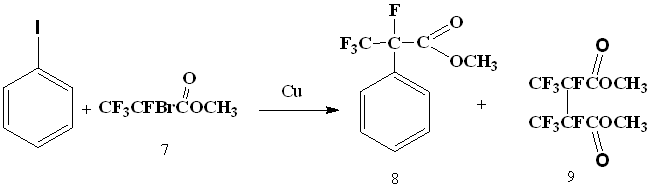
The next step was an attempt to introduce 2-bromotetrafluoropropionic acid methyl ester (7) into the reaction with iodobenzene.
It turned out that methyl ester of 2-bromo-tetrafluoropropionic acid in absolute dimethylformamide reacts with iodobenzene, under the influence of metallic copper, resulting in formation of methyl ester of α-phenyltetrafluoropropionic acid (8). The best yield was obtained using 2.5 equivalents of metallic copper and carrying out the reaction at 115-120°C.
The reaction runs for 2 h at 60°C, for 20 minutes - at 90°C and almost instantly - at 110-120°C. At lower temperatures, the main reaction product becomes the product of 2-bromotetrafluoropropionic acid methyl ester doubling (9). At higher temperatures, a significant formation of 2H-tetrafluoropropionic acid is observed, apparently due to the reaction of copper derivative of bromodifluoroacetic acid with a solvent. The formation of biphenyl under these conditions was not observed.
Then salt (10) was obtained from α-phenyltetrafluoropropionic acid methyl ester (8) and decarboxylated in N-methylpyrrolidone to give the trifluorostyrene (4).
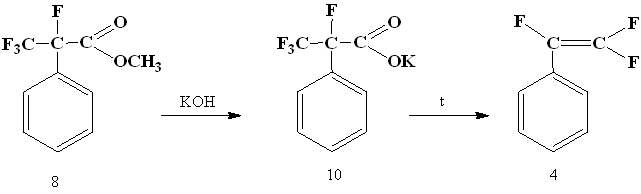
Thus, we have developed a new synthesis method of trifluorostyrene from available reagents. does not require the special equipment for its implementation. The total yield of trifluorostyrene was 39%.
Experimental part
1H and 19F NMR spectra were recorded via Bruker AVANCE-300 spectrometer at 300 and 282 MHz, respectively, using external standard CDCl3. Chemical shifts for 1H NMR spectra are given in ppm relatively to TMS. Chemical shifts of the 19F NMR spectra are given in m. d. relatively to CFCl3.
2-Bromo-tetrafluoropropionic acid methyl ester 7 was obtained as described in [21]. For absolutization of solvents the standard ways and operation methods was used.
Preparation the methyl ester of α-phenyltetrafluoropropionic acid
600 ml of absolute DMF, 314 g (1.54 mol) of iodobenzene and 244 g (3.84 mol) of copper powder were heated to 110-115°C with stirring, 330 g (1.38 mol) of 2-bromotetrafluoropropionic acid methyl ester were added dropwise during 1 h, maintaining the temperature in the range of 115‑120°C. The reaction mass was kept at 120°C for 2 h, cooled to room temperature, filtered from copper salts at a Nutsche filter. Obtained filtrate was transferred to a distillation flask, 300 ml of water was added and water raw solution, containing the initial iodobenzene, the product doubling of 2,3,3,3‑tetrafluoro-2-bromopropionic acid methyl ester, 2H-tetrafluoropropionic acid methyl ester and α‑phenyltetrafluoropropionic acid methyl ester, was distilled at 100°C. The lower layer was separated and fractionated under reduced pressure, collecting a boiling fraction at 62-70°C and pressure 2 Torr. 228 g of α-phenyltetrafluoropropionic acid methyl ester was obtained as a colorless liquid with a purity of 84% GC, which was used at the next stage without further purification. Calculated yield - 58%.
1H NMR, δ.: 3.6 (s, 3H, CH3) 7.2 (m, 3Н C6H5), 7.5 (s 2Н C6H5)
19F NMR, δ.: -173.3 (m, 1F, CF), -78.1 (s, 3F, CF3).
Preparation of potassium α-phenyltetrafluoropropionate
Potassium hydroxide 56 g (1 mol) was dissolved in 200 ml of methanol, 228 g (84%) (0.81 mol) of α-phenyltetrafluoropropionic acid methyl ester was added dropwise at 60°C, boiled during 1 h, then poured into a porcelain cup, dry evaporated, washed with diethyl ether and then dried in a flask in a boiling water bath at <0.5 Torr to constant weight. 147 g (0.56 mol) of potassium α‑phenyltetrafluoropropionate were obtained as a white powder.
1H NMR, (D2O) δ: 7.3 (m, 3H C6H5), 7.6 (m, 2H C6H5);
19F NMR, δ: -161 (s 1F, CF), -76 (s 3F, CF3).
Preparation of 1,2,2-trifluorostyrene
97 g (0.37 mol) of potassium α-phenyltetrafluoropropionate was dissolved in 120 ml of abs. N-methylpyrrolidone and 1g TEMPO was added. The reaction mass was heated at pressure 60 Torr created by adjustable water jet pump until the start of decarboxylation at 110-115°C. During decarboxylation, this pressure was reduced to 120 Torr. After 30 min decarboxylation ceased the pressure returned to 60 Torr, then this pressure was adjusted to 30 Torr, which resulted in distillation of the product into a cooled receiving flask. 56 g of trifluorostyrene were obtained as a colorless liquid with GC purity of 97.5%.
1H NMR, δ: 7.1 (m, 3H, C6H5), 7.25 (m, 2H, C6H5)
19F NMR, δ: -102.7 (d. d., 2JFF =71 Hz, 3JFF =30 Hz, CFC=CFACFB), -117.2 (d. d., 2JFF =70 Hz, 3JFF =108 Hz, CFC=CFACFB). -178.6 (d. d., 3JFF =109Hz, CFC=CFACFB).
Acknowledgments
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (Contract/agreement No. 075-000697-22-00) and was performed employing the equipment of Center for molecular composition studies of INEOS RAS.
References
- Patent CN114210532, 2022.
- Patent CN113786743, 2021.
- Patent US7550216, 2009.
- Patent CN101759832, 2012.
- Patent US10556973, 2018.
- Patent CN105977433, 2016.
- Nikitina T. S., Advances in Chemistry, 1990, v. 59, 995-1020 (in Russian); Fu Yong-zhu, Liu Fu-Qiang, Xing Dan-Min, Ju Jing-rong, Yi Bao-lian, Zhang Hua-Min, Dianyuan jishu, Chin. J. Power Sources, 2003, 27(4), 345-347
- Cohen S.G, Wolosinski H.T, Scheuer P.J., J. Am. Chem. Soc., 1949; 71, 3439-3440
- Dixon, S. J., Org. Chem., 1956, 21, 400.
- Heinze P.L. and Burton D.J., J. Org. Chem., 1988, 53, 2714-2720.
- Qibo L., Burton D.J., Journal of Fluorine Chemistry, 2011, 132(2), 78-87.
- Sorokina R. S, Rybykova L. F., Kalinovsky I. O., Chernoplekova V. A., Beletskaya. I.P., Zh. Org. Kh., 1982, 18(11), 2458-2459 (in Russian).
- Heinze P.L, Burton D.J., J. Fluorine Chem., 1986, 31(1), 115-119.
- Hansen S.W., Spawn T.S., Burton D.J., J. Fluorine Chem., 1987, 35(2), 415-420.
- R. Anilkumar, Donald J. Burton, Tetrahedron Letters, 2003, 44(35), 6661-6664.
- Ohashi M., Kambara T., Hatanaka T., Saijo H., Doi R., Ogoshi S., Journal of the American Chemical Society, 2011, 133 (10), 3256-3259.
- Saijo, Hiroki; Sakaguchi, Hironobu; Ohashi, Masato; Ogoshi, Sensuke,Organometallics, 2014, 33(14), 3669-3672.
- Masato Ohashi, Hiroki Saijo, Mitsutoshi Shibata; Sensuke Ogoshi, European Journal of Organic Chemistry, 2013, 3, 443-447.
- Becker R. A., Polyfluorine-containing metastable enols: Dissertation for degree of Doctor of Chemical Sciences, Moscow, 1979, 130-132 (in Russian).
- Patent US6350925, 2002.
- Syntheses of organofluorine compounds, Part 2, edited by S.M. Igumnov, E.V. Igumnova, M., "Trovant", 2011, 351p., 211-212 (in Russian).
ARTICLE INFO
Received 22 July 2022
Accepted 1 August 2022
Available online August 2022
Recommended for publication by PhD Olga Bryzgalova
eLIBRARY Document Number (EDN) YGQDHA

Fluorine Notes, 2022, 143, 5-6
