Received: September 2023
DOI 10.17677/fn20714807.2023.05.02
Fluorine Notes, 2023, 150, 3-4
SYNTHESIS OF FLUORINE- OR CHLORINE CONTAINING n-PYRIDINYLOXY-SUBSTITUTED BENZAMIDE OXIMES
T. P. Vasilyeva*, D. V. Vorobyova
A.N. Nesmeyanov Institute of Organoelement Compounds RAS, Russian Federation, 119334, Moscow, Vavilov str. 28/1.
Fax: +7(499) 135 5085. E-mail: d-20@mail.ru
Abstract: New 4-((3,5-dichloropyridin-4-yl)oxy)benzonitrile 2 was obtained by a model reaction of trichloropyridine 1 with 4-cyanophenol in the presence of K2CO3 in DMF. Under similar conditions, the action of 4-cyanophenol to 2-chloro-5- (trifluoromethyl)pyridine previously difficult to obtain 4-((5-trifluoromethyl)pyridin-2-yl)-oxy)benzonitrile 4 was synthesized in high yield. Based on the obtained nitriles 2 and 4, the preparative methods for synthesis of new fluorine- or chlorine-containing n-pyridinyloxy-substituted benzamidoximes 3 and 5 have been developed, that suitable for scaling.
Keywords: 3,4,5-trichloropyridine, 4-cyanophenol, 2-chloro-5-(trifluoromethyl)pyridine, fluorine- or chlorine-containing n-pyridyloxy-substituted benzonitriles and benzamidoximes, hydroxylamine.
Previously, we developed the method for preparation of fluorinated benzamides [1]. This article is devoted to other derivatives – n-pyridyloxy-substituted benzonitriles and amidoximes. It is known that key fragments of diaryl ethers (including those containing a pyridine ring) appear in some insecticides (pyriproxyfen, fenoxycarb) and herbicides (fusilade, chlometoxynil) [2]. In contrast to benzonitriles, the benzamidoximes (N-hydroxy-benzimidamides) are a poorly studied class of compounds with potential biological activity. Thus, the high antimalarial activity of amidoxime 3 was reported [3], but this study does not contain a method for its synthesis and physical characteristics. We supposed that a convenient starting compound for preparation of both substance 3 and the corresponding nitrile 2 would be readily available 3,4,5‑trichloropyridine 1 (see Fig. 1).
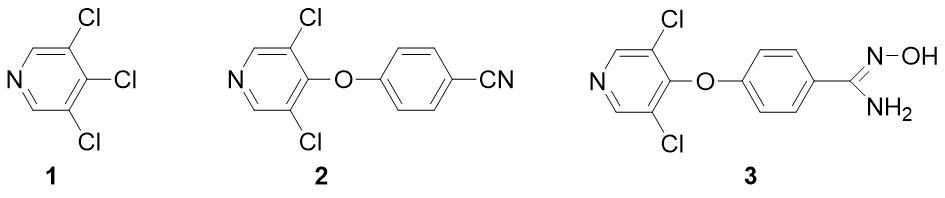
Figure 1.
The goal of this study is to develop the preparative syntheses of compounds 2 and 3 and use these methods to obtain CF3-containing analogues (nitrile and amidoxime). It is known that fluorine-containing drugs have increased activity compared to hydrogenated analogues [4, 5]. Before our study, 3- or 4-pyridyloxy-substituted benzonitriles were prepared by reaction of 4- or 2fluoropyridines with cyanophenols in DMF in the presence of Et3N [2] or potassium carbonate [6] (see Scheme 1).
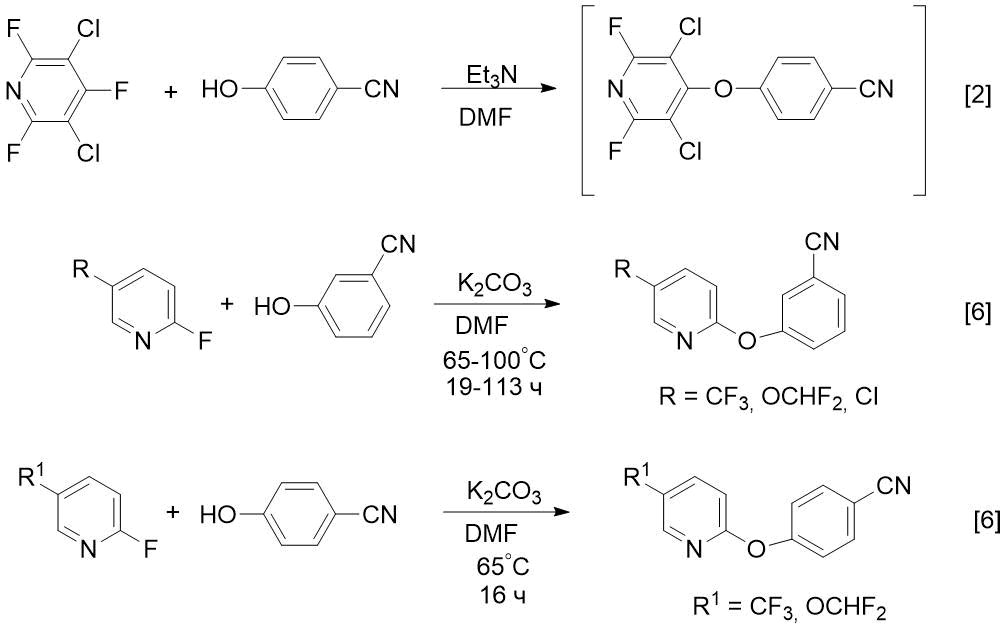
Scheme 1. Known methods for preparing m- or p-pyridyloxy-substituted benzonitriles from 4- or 2-fluoropyridines [2, 6].
The disadvantage of these methods is the high cost of fluoropyridines (for example, 2‑fluoro-5-trifluoromethylpyridine). We have developed a more economical method for synthesis of such nitriles based on available chloropyridines, in particular, trichloropyridine 1. Previously, 1 was obtained in low yield (along with other products) by reactions of pyridine [7] or α-picoline [8] with chlorine. It is also known about synthesis of 1 by interaction of PCl5 and POCl3 with 3,5-dichloro-4-pyridone (without indicating the yield) [9], however, in this case, due to aggressive reagent (PCl5) and high temperature (125°C), the asphaltization of reaction mixture occurred. According to our data, the optimal method for obtaining 1 is interaction of 3,5-dichloro-4-pyridone with a double excess of POCl3 in the presence of catalytic amounts of dimethylformamide and moderate heating. By reacting trichloropyridine 1 obtained in this way with 4-cyanophenol in the presence of K2CO3, we were able to obtain the target nitrile 2 with high yield; in this case, the chlorine atom was replaced in position 4 of pyridine cycle (see Scheme 2).
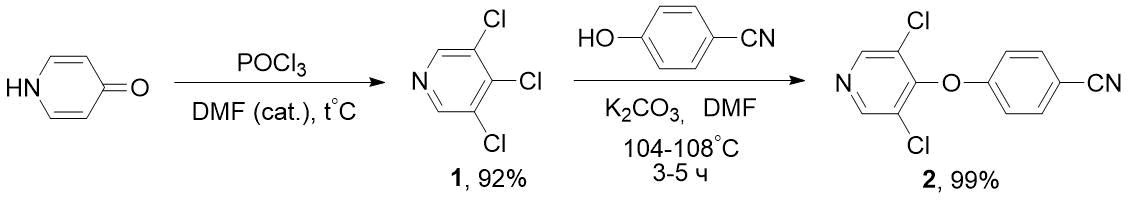
Scheme 2. Synthesis of 4-((3,5-dichloropyridin-4-yl)oxy)-benzonitrile 2 from 3,4,5-trichloropyridine 1.
To obtain F-containing pyridyloxybenzonitrile 4, we used 2-chloro-5-(trifluoromethyl)pyridine as starting compound, which is much lower in cost than its 2-fluorine analogue. This synthesis was carried out similarly to preparation of nitrile 2, but under more stringent conditions - at 103-128°C for 8-10 h (see Scheme 3). Nevertheless, 2-Cl-5-CF3-pyridine turned out to be more reactive than 2-F-analogue, for which the reaction time was 16 h [6] (see Scheme 1).
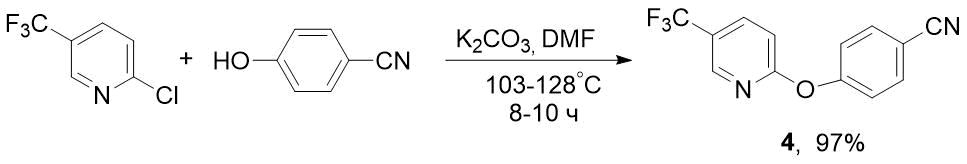
Scheme 3. Synthesis of 4-((5-(trifluoromethyl)pyridin-2-yl)oxy)benzonitrile 4 from 2-chloro-5-(trifluoromethyl)pyridine.
Pyridyloxy-substituted benzamidoximes were previously obtained by heating a mixture of corresponding nitriles, hydroxylamine hydrochloride and sodium bicarbonate in aqueous-alcoholic medium, but the resulting target products were used in synthesis of drugs without purification [6] (see Scheme 4).
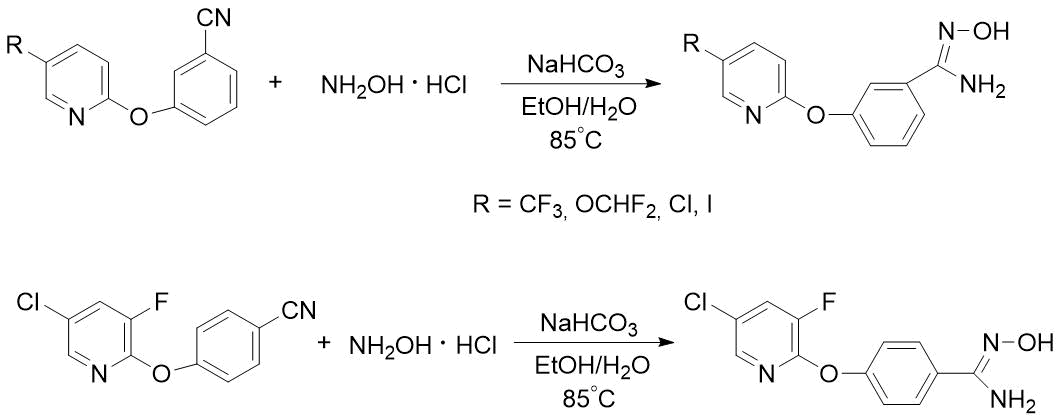
Scheme 4. Syntheses of m- or n-pyridyloxy-substituted benzamidoximes described in [6].
Using the example of preparation of heterocyclic amidoximes 3 and 5, we have shown that purer products are formed when the process is carried out in two stages, i. e. 1) in situ generation of hydroxylamine by the action of sodium carbonate to NH2OH·HCl in water; 2) adding the solution of NH2OH to nitriles 2 or 4 in alcohol, followed by heating (see Scheme 5).
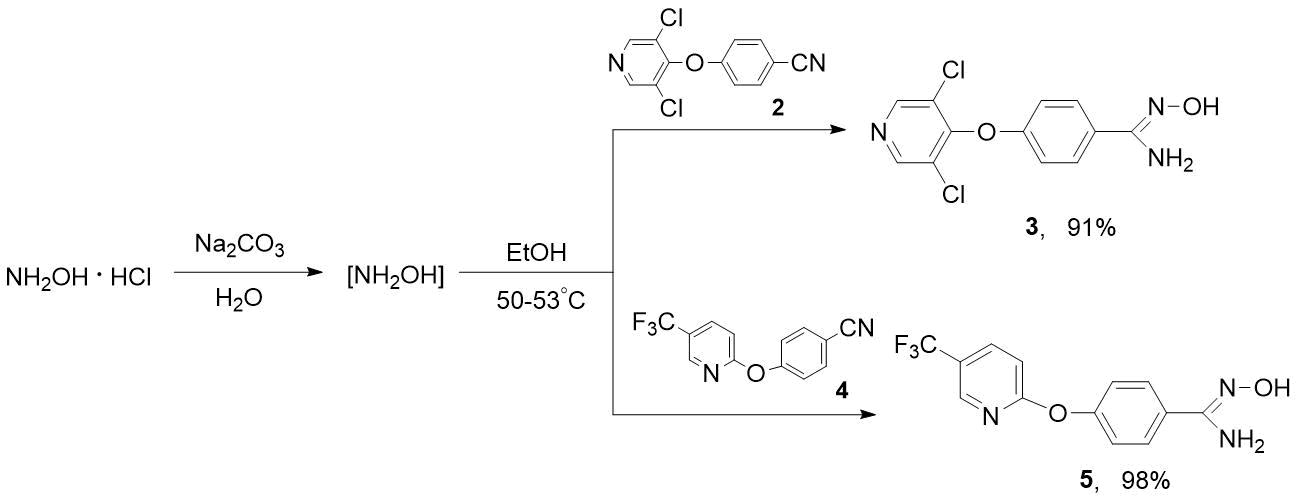
Scheme 5. Synthesis of heterocyclic amidoximes 3 and 5.
It is obvious that this developed approach to synthesis of compounds 3 and 5 can be successfully applied to obtain other polyfluoro- and polyfluoroalkyl-containing amidoximes. The synthesized amidoximes 3 and 5 are the crystalline solids that can be purified by reprecipitation from solution in aqueous sodium bicarbonate by adding 2N hydrochloric acid. The composition and structure of these compounds have been proven by NMR spectroscopy and elemental analysis. In 1Н NMR spectra 3, 5, singlets of hydroxyl protons (=N-OH) at 9.62-9.67 ppm and amino groups (NH2) at 5.80-5.86 ppm are characteristic. The signal of CF3 group of amidoxime 5 in 19F NMR spectrum is observed at -60.04 ppm. In mass spectrum 5, the molecular ion is basic.
Thus, as a result of this study, on the basis of 2-chloro-5-(trifluoromethyl)pyridine, the economical synthesis of previously difficult-to-find nitrile 4 with high yield has been developed. This compound is important object with pharmacophore core and is used in synthesis of triazine inhibitors of angiogenesis (for treatment of cancer and macular degeneration of eyes) [10], heteroaryl derivatives of aminopropanol, used for treatment of asthma, allergies and atherosclerosis [6], as well as derivatives 1,2, 4-oxadiazoles, proposed for treatment of autoimmune and vascular diseases [11]. As for synthesized amidoximes, since the dichloro-substituted compound 3 turned out to be biologically active [3], the new CF3-containing analogue 5 we obtained is certainly no less interesting for medicinal chemistry than original nitrile 4. This is confirmed by the fact that some benzamidoximes containing in the n- or m‑position, substituted (pyridin-2-yl)oxy groups are often used to obtain medicinal substances [6].
Experimental part
NMR spectra were recorded via Bruker WM-250 and Bruker Avance-300 spectrometers with operating frequencies of 250 MHz and 300 MHz for 1Н NMR (using internal standard SiMe4) and 282 MHz for 19F NMR (using internal standard CFCl3), respectively. Elemental analysis was carried out in the Laboratory of Microanalysis of INEOS RAS. The progress of reactions was monitored by TLC at Merck plates (silica gel 60 F254, 0.25 mm). The RF values of obtained compounds were determined in the systems acetone/ СCl4 = 1:10 or in the systems ethyl acetate/petroleum ether = 2:1. The starting materials 3,5-dichloro-4-pyridone, 2-chloro-5-(trifluoromethyl)pyridine and 4-cyanophenol are commercially available reagents. All used solvents were purified using standard methods.
3,4,5-Trichloropyridine (1)
4.2 ml of DMF was added dropwise to a mixture of 46.0 g (0.28 mol) of 3,5-dichloro-4-pyridone and 86.3 g (0.56 mol) of phosphorus oxychloride. After stirring at 20°C for 1 h the reaction mixture was refluxed until the reaction was complete (3-5 h), then poured into a glass of ice water (650 ml), and then this mixture was neutralized to pH ~ 5-7 by adding 40% -NaOH (139 ml). The precipitate that was filtered off and washed three times with cold water and then- with cold aqueous alcohol (1:1) (80 ml). Obtained product was dried in air, then - in vacuum: first - over CaCl2, then - over P2O5. Obtained 47.09 g (92%) of compound 1 in the form of light beige powder with m.p. 78-79°C (in [7] 74-75°C, in [8] – at 73-74°C, in [9] – at 76-77°C, RF (solution in CHCl3) 0.82 (in the system of CHCl3/MeOH = 37:3); 0.68 (in the system of acetone/CCl4 = 1:10).
1Н NMR spectrum (300 MHz, CDCl3, δ, ppm): 8.53 (s, 2H, 2CH).
4-((3,5-Dichloropyridin-4-yl)oxy)benzonitrile (2)
To a solution of 46.8 g (0.26 mol) of trichloropyridine 1 and 30.52 g (0.26 mol) of 4‑cyanophenol in 210 ml of DMF, 42.47 g (0.31 mol) of crushed К2CO3 was added, then the reaction mixture was heated with stirring at 104-108°C until this reaction is complete (3-5 h). The contents of flask were cooled and poured into a glass with ice water (1320 ml). The resulting precipitate was filtered off, washed three times with cold water, and then dried in air. Obtained 67.0 g (99%) of compound 2 in the form of light crystals with m.p. 141-143°C and RF = 0.35 (solution in CHCl3, in the system of acetone/CCl4 = 1:10).
1Н NMR spectrum (250 MHz, CDCl3, δ, ppm, J/Hz): 6.91 (d, 2H, Ar, J = 9.0); 7.66 (d, 2H, Ar, J = 9.0); 8.61 (s, 2H, 2CHPy).
Founded (%): C, 54.51; N, 2.26; N, 10.59. C12H6Cl2N2O.
Calculated (%): C, 54.34; N, 2.26; N, 10.57.
4-((3,5-Dichloropyridin-4-yl)oxy)-N'-hydroxybenzimidamide (3)
Stage 1. Preparation of solution of hydroxylamine in water. To a solution of 26.77 g (0.25 mol) of Na2CO3 in 102 ml of warm (~42°C) water, 35.14 g (0.5 mol) of hydroxylamine hydrochloride was gradually added and stirred at this temperature until the evolution of CO2 ceased.
Stage 2. A warm solution of NH2OH (according to Stage 1) was added to a suspension of 66.93 g (0.25 mol) of nitrile 2 in 1100 ml of ethanol at 43°C with stirring; then the resulting mixture was heated at 51-53°C until the reaction was completed (6-7 h, TLC control in the system of ethyl acetate/petroleum ether = 2:1). The reaction mixture was cooled to 0÷5°C, then resulting precipitate was filtered off and washed with cold solvents: first - with water (65 ml x 3 times), then - with methanol (2 x 55 ml) and petroleum ether (2 x 45 ml). Obtained product was dried in air and in vacuum - first - over CaCl2, then - over P2O5. Obtained 68.46 g (91%) of compound 3 in the form of white powder, m.p. 224-225°C and RF (solution in warm methanol) = 0.27-0.35 (in the system of ethyl acetate/petroleum ether = 2:1); 0.59 (in ethyl acetate).
1Н NMR spectrum (300 MHz, d6-DMSO, δ, ppm, J /Hz): 5.80 (s, 2H, NH2); 6.93 (d, 2H, Ar, J = 8.6); 7.66 (d, 2H, Ar, J = 8.6); 8.79 (s, 2H, 2CHPy); 9.62 (s, 1H, =N-OH).
Founded (%): C, 48.11; N, 2.87; N, 13.99. C12H9Cl2N3O.
Calculated (%): C, 48.32; N, 3.02; N, 14.09.
4-((5-(Trifluoromethyl)pyridin-2-yl)oxy)benzonitrile (4)
This synthesis was carried out similarly to preparation of nitrile 2 from 89.58 g (0.49 mol) of 2-chloro-5-(trifluoromethyl)pyridine, 58.73 g (0.49 mol) of 4-cyanophenol and 81.73 g (0.59 mol) К2CO3 in 280 ml of DMF, but with more longer heating (8-10 h), gradually increasing the bath temperature from 103°C to 128°C - until cyanophenol disappears (TLC control in the system of acetone/ CCl4 = 1:10). The product was washed with water and cold petroleum ether (100 ml), dried in air, then in vacuum over CaCl2. We obtained 126.4 g (97%) of compound 4 in the form of white crystals, m.p. 85-87°C (from EtOH) and RF = 0.49 (solution in CH2Cl2, in the system of acetone/CCl4 = 1:10).
1H NMR spectrum (cf. [6]) (300 MHz, СDCl3, δ, ppm, J /Hz): 7.11 (d, 1H, СНPy, J = 7.1); 7.24-7.29 (m, 2H, Ar); 7.71 (d, 2H, Ar, J = 8.7); 7.96 (d.d., 1H, СНPy, J = 8.7, J = 1.8); 8.41 (s, 1H, СНPy).
N'-hydroxy-4-((5-trifluoromethyl)pyridin-2-yl)oxy)benzimidamide (5)
It prepared similarly to the synthesis of amidoxime 3. A solution of hydroxylamine (from 23.65 g, 0.34 mol NH2OH*HCl and 18.04 g, 0.17 mol Na2CO3 in 68 ml of water) was added to solution of 63.7 g (0.24 mol) nitrile 4 in 980 ml of ethanol at 41-42°C, then the reaction mixture was stirred at 50°C for 5-6 h. The temperature of this mixture was brought to 20°C, the formed precipitate (NaCl) was filtered off and washed with methanol (2 x 50 ml). The mother liquor was evaporated in vacuum to a volume of ~250 ml, obtained residue was cooled and mixed with cold water (500 ml). The resulting precipitate was filtered off, washed with cold water (2 x 80 ml) and dried: first - in air, then - in vacuum (over CaCl2 and P2O5). We obtained 70.2 g (98%) of compound 5 in the form of shiny white crystals, m.p. 138-141°C (after purification by reprecipitation of NaHCO3 from a solution in 2N hydrochloric acid, m.p. 140-143°C) and RF (solution in MeOH) = 0.45 (in the system of ethyl acetate/petroleum ether = 2:1).
1H NMR spectrum (300 MHz, d6-DMSO, δ, ppm, J/Hz): 5.86 (s, 2H, NH2); 7.21 (d, 2H, Ar, J = 8.4); 7.27 (d, 1H, CHPy, J = 7.8); 7.74 (d, 2H, Ar, J = 8.4); 8.24 (d, 1H, СНPy, J = 7.8); 8.57 (s, 1H, СНPy), 9.67 (s, 1H, =N-OH).
19F NMR spectrum (282 MHz, d6-DMSO, δ, ppm): -60.04 (s, 3F, СF3).
Mass spectrum, m/z: 297 [M+], 100%.
Founded (%): C, 52.59; N, 3.31; N, 14.04. C13H10F3N3O2.
Calculated (%): C, 52.53; N, 3.37; N, 14.14.
Acknowledgements
This work was supported by the Ministry of Science and Higher Education of the Russian Federation (Contract/agreement No. 075-03-2023-642) and was performed employing the equipment of Center for molecular composition studies of INEOS RAS.
References
- Vasilyeva T.P., Dyachenko V.I., Vorobyeva D.V., Fluorine notes, 2022, 143(4), 3-4.
- Litvak V.V., Mainagashev I.Ya., Bukhanets O.G., RAS Chem. Bull., 2007, 56, 980.
- Rastelli G., Pacchioni S., Sirawaraporn W., Sirawaraporn R., Parenti M., Ferrari A., J. Med. Chem., 2003, 46, 2834.
- Ricci G., Ruzziconi R., J. Org. Chem., 2005, 70, 611.
- Zhu Y., Han J., Wang J., Shibata N., Sodeoka M., Soloshonok V.A., Coelho J., Toste F., Chem. Rev., 2018, 118, 3887.
- Kordikowski A., Liu Y., Lüӧnd R., Markert C., Miltz W., Roehn T., Spanka C., Thoma G., Xie T., WO2022219546A1, 2022.
- Wibaut J., Nicolai J., Rec. Trav. Chem. Pays-Bas, 1939, 58, 709.
- Shlenkova E.K., Kuprianova N.S., Butkova O.L., Katzobashvili V. Ya, Bear A.A., J. Org. Chem. 1977, 13, 446.
- Dohrn M., Diedrich P., Justus Liebigs, Ann. Chem., 1932, 494, 284.
- Henkin J., Davidson D., Sheppard G., Woods K., McCroskey R., WO9931088, 1999.
- DAS, Jagabandhu, KO, Soo Sung, WO2012040532, 2012.
ARTICLE INFO
Received 14 September 2023
Accepted 12 October 2023
Available online October 2023
Recommended for publication by PhD M.A. Manaenkova
eLIBRARY Document Number (EDN) UYKOHU

Fluorine Notes, 2023, 150, 3-4
