Received: June 2024
DOI 10.17677/fn20714807.2024.03.02
Fluorine Notes, 2024, 154, 3-4
SYNTHESIS OF 5-FLUOROSULFONYLPERFLUOROPENTYL VINYL ETHER – A NEW MONOMER FOR PERFLUOROSULFOACIDIC IONOMERS
A.A. Tyutyunov,a,b,c G.А. Khromov,a S.М. Igumnova,b
aA.N. Nesmeyanov Institute of Organoelement Compounds of the Russian Academy of Sciences, 28 Vavilova St., Build. 1, 119334 Moscow, Russian Federation
bP@M-Invest Scientific Production Association, 42 Vavilova St., Build. 1-2-3, 119119 Moscow, Russian Federation
cHydrogen Energy Center LLC
3 Akad. Semenova prosp., Office 3, 142432 Moscow Region, Chernogolovka, Russian Federation
e-mail: tuytuynov@rambler.ru
Abstract: Perfluoroallyl iodide undergoes thermally-induced addition to tetrafluoroethylene already at 70 – 90C to give mainly 5-iodoperfluoropentene, which is a key intermediate for the synthesis of a new fluorosulfonylperfluoropentyl vinyl ether, which is of interest as a monomer for the preparation of perfluorosulfonic acid proton exchange membranes.
Key words: fluorosulfonylperfluorovinyl ethers, monomers, perfluorosulfonic acid membranes.
Intensive development of fluoropolymer chemistry at DuPont in the mid-1960s led to the creation of perfluorosulfonic acid ionomers based on copolymers of tetrafluoroethylene with perfluoro-2-(2-fluorosulfonylethoxy)propyl vinyl ether, which were produced under the trade name Nafion and used as proton exchange membranes in various electrochemical processes, including hydrogen fuel cells [1]. Subsequent research by Dow Chemical, Solvay Specialty Polymers, Asahi Glass, Asahi Kasei, and 3M Fluoropolymers led to the production of similar Nafion-like perfluorosulfonic acid polymers using short- and medium-chain fluorosulfonyl perfluoroalkyl vinyl ethers [2-4].
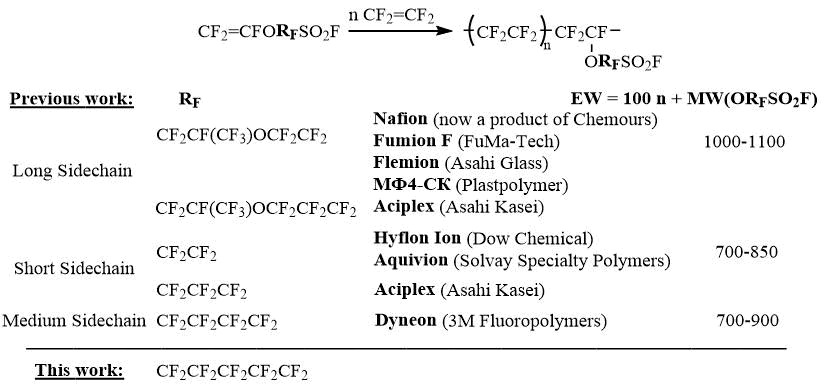
Numerous comparative studies of the properties of these proton exchange membranes over the last few decades have shown that there is a complex relationship between the molecular structure and morphology of the ionomer, consisting of different phases of crystalline (similar to PTFE) and ionic (hydrated sulfonic acid) domains, and its transport, mechanical, and other characteristics [5-6]. Perfluorosulfonic acid membranes based on short- and medium-chain fluorosulfonyl perfluoroalkyl vinyl ethers have been preferred for efficient use in fuel cells [7-8]. These membranes have lower equivalent weight (EW) (higher ion exchange capacity), which increases the capacity of the fuel cell. They can operate at higher temperatures and lower humidity, which simplifies the cooling system and increases the catalyst's resistance to fuel contamination. However, all such perfluorosulfonic acid membranes require additional modifications to increase membrane stability in the oxidizing environment of the hydrogen cell and to extend their service life [9].
These data indicate that further elongation of the perfluoroalkyl moiety in the fluorosulfonyl monomer should increase the equivalent mass of the perfluorosulfonic acid membrane and compromise its performance. However, in the case of the monomer CF2=CFO(CF2)5SO2F, compared to the same monomer from the 3M Company, the ionomer EWs increase by an average of only 5%, and thus the synthetic availability of a particular monomer for perfluorosulfonic acid membranes remains the decisive argument. Therefore, the aim of this work was to develop a preparative method for the synthesis of a new 5-fluorosulfonylperfluoropentyl vinyl ether for further studies of the properties of polymers derived therefrom [10-11].
In contrast to perfluoro-2-(2-fluorosulfonylethoxy)propyl vinyl ether, which is used for the production of Nafion membranes and which is accessible via almost the only synthetic protocol [1], several approaches have been developed for the short-chain monomers CF2=CFO(CF2)nSO2F, n = 2, 3, but most of them require the use of hard-to-reach reagents such as 3-chloropentafluoropropene-1,2-oxide or molecular fluorine, and complex equipment [2]. At the same time, we have developed an affordable technology for the preparation of CF2=CFOCF2CF2SO2F based on the sulfinatodehalogenation of perfluorobromoethyl vinyl ether [12]. In turn, the 3M monomer CF2=CFO(CF2)4SO2F can be obtained either by fluorination (ECF or F2/N2) of various organic precursors or by autoclave oxidation of 4-fluorosulfonylperfluorobutene, which is only feasible at a specialized production facility [13-14].
For the synthesis of the monomer CF2=CFO(CF2)5SO2F, it is most rational to use industrially available fluoroolefins as organofluorine precursors. Thus, perfluoroallyl iodide (1) can be obtained from hexafluoropropylene according to the known procedure, which can be implemented in practice without any difficulties [15].
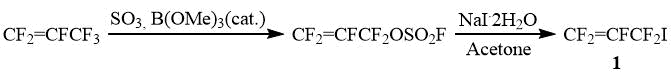
Although perfluoroallyl iodide (1) has been known for a relatively long time, its reactions with tetrafluoroethylene have not been reported. We have found that 1 undergoes thermally-induced addition to CF2=CF2 at 7090C to give mainly 5-iodoperfluoropentene (2) and minor amounts (57%) of perfluoro-1,5-hexadiene and 1,2-diiodotetrafluoroethane. Further conversion of compound 2 to the target monomer can be accomplished by the following standard chemical transformations.
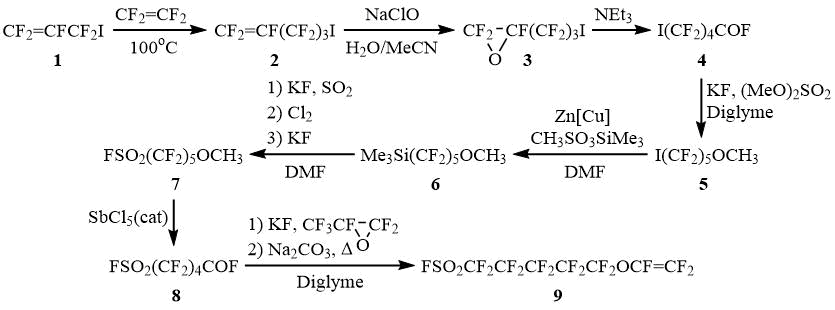
It should be noted that despite the multistep nature of this synthesis, these reactions do not require the use of complex equipment or hazardous and hard-to-reach reagents. In addition, the intermediates formed in certain steps are of particular practical interest and can serve as starting materials for the preparation of various commercially demanded organofluorine compounds. Thus, the product of the next to the last step, FSO2(CF2)5OCF(CF3)COF, can be used to prepare perfluorinated surfactants, further extending the utility of compounds synthesized according to the said scheme.
Experimental part
1H, 19F NMR spectra were recorded on a Bruker AM-300 spectrometer with 300 and 282 MHz operating frequencies, respectively. Chemical shifts in 1H NMR spectra were referenced to the residual proton chloroform peak (7.26 ppm in CDCl3) and reported in ppm units relative to TMS. Chemical shifts in 19F NMR spectra are reported in ppm units relative to an external CFCl3 reference. Positive values of chemical shift correspond to the downfield shift of the indicator nucleus signal.
Perfluoroallyl fluorosulfate was obtained by standard procedure from hexafluoropropene [15].
Perfluoroallyl iodide (1)
To a vigorously stirred slurry of NaI*2H2O (223 g, 1.2 mol) in acetone (750 mL), CF2=CFCF2OSO2F (250 g, 1.09 mol) was added at such a rate that the temperature of the mixture is kept in the range of 20-25°C. Next, the reaction mixture was stirred at 25-30°C for 3 h, poured into an equal volume of ice water, the bottom layer was separated, washed twice with an equal volume of ice water and distilled from an equal volume of H2SO4 (conc.), collecting the fraction boiling at 54 – 56°C to give CF2=CFCF2I (225 g, yield 80%).
19F NMR, δ : -49.2 (ddd, 5 Hz, 21,5 Hz, 32 Hz, 2F, ICF2), -97 (tdd, 5 Hz, 38 Hz, 54 Hz, 1F, CF=CFcisFtrans), -106.1 (tdd, 32 Hz, 54,5 Hz, 116 Hz, 1F, CF=CFcisFtrans), -178.6 (tdd, 21 Hz, 38 Hz, 116 Hz, 1F, CF=CFcisFtrans).
5-Iodoperfluoropentene (2)
A steel autoclave was loaded with CF2=CFCF2I (100 g, 0.39 mol), closed, cooled with liquid nitrogen for 5 minutes, and evacuated to a residual pressure of 0.1 – 0.5 Torr. Then, the autoclave valve was closed, a rubber bag containing CF2=CF2 (~10 L, ~0.44 mol) was connected thereto, the autoclave valve was opened and CF2=CF2 was condensed into the autoclave. The autoclave was then sealed and heated on a mechanical shaker to 90 – 100°C for 10 hours. After that, the autoclave was cooled to room temperature, opened and the resulting mixture was distilled. The solvent strip was collected containing a mixture of unreacted CF2=CFCF2I and by-product CF2=CFCF2CF2CF=CF2 (bp 60C), which can be recycled, and a product with bp 95 – 102°С, which is CF2=CF(CF2)3I with an admixture of ~20% ICF2CF2I (bp 112-113°С; 19F NMR, δ : -54 (s)).
CF2=CF(CF2)3I (105 g, yield 60%, ~80% purity) was obtained, which was used in the subsequent step without further purification.
19F NMR, δ : -59.8 (dt, 1.5 Hz, 3 Hz, 2F, ICF2), -90.5 (tdd, 6 Hz, 40 Hz, 52 Hz, 1F, CF=CFcisFtrans), -106.8 (tdd, 27 Hz, 52 Hz, 117,5 Hz, 1F, CF=CFcisFtrans), -116.3 (d, 2 Hz, 2F, ICF2CF2), -118.1 (dt, 4 Hz, 10.5 Hz, 2F, ICF2CF2CF2), -189.8 (ddd, 17 Hz, 22 Hz, 40 Hz, 1F, CF=CFcisFtrans).
5-Iodoperfluoropentene oxide (3)
Chlorine (194 g, 2.73 mol) was bubbled into a vigorously stirred solution of NaOH (241 g, 6 mol) in water (960 mL) at -25 ÷ -20°С. After that, CF2=CF(CF2)3I (466 g, 1.3 mol based on the pure product) containing ~20% ICF2CF2I, and acetonitrile (300 mL) were added to the resulting aqueous solution of NaClO. Vigorous stirring was continued, the mixture was heated to -5 ÷ 2°C, at which point the exothermic reaction begins, during which the temperature of the reaction mixture was kept within 5 – 10°C by means of a cooling bath. The reaction mixture was stirred for one hour at ~15°С and diluted with an equal volume of water. The bottom layer was separated, washed with an equal volumes of water, 5% hydrochloric acid and distilled over P2O5, collecting the fraction boiling at 90-100°С to give 5-iodoperfluoropentene oxide (371 g, yield 61%) containing ~20% ICF2CF2I. The product was used in the subsequent step without further purification.
19F NMR, δ : -63.3 (s, 2F, ICF2), -110.4 (ddd, 13 Hz, 30 Hz, 42 Hz, 1F, CFCFcisFtransO), -113.4 (dd, 18 Hz, 42 Hz, 1F, CFCFcisFtransO), -114.7 (dd, 13 Hz, 289 Hz, 1F, ICF2CFAFB), -116 (dd, 9 Hz, 292 Hz, 1F, ICF2CFAFB), -119.5 (dq, 13.5 Hz, 41 Hz, 1F, ICF2CF2CFAFB), -124.6 (dd, 10 Hz, 293 Hz, 1F, ICF2CF2CFAFB), -152.3 (dd appears as t, 16 Hz, 1F, CFCFcisFtransO).
5-Iodoperfluoropentanoyl fluoride (4)
To stirred NEt3 (5.4 g, 0.05 mol), 5-iodoperfluoropentene oxide (500 g, 1.07 mol based on the pure product) containing ~20% ICF2CF2I was added dropwise, which raised the temperature of the reaction mixture to 80 – 90°C. The mixture was heated to reflux for 1 hour and distilled, the distillate boiling at 90 – 100°C was collected to give I(CF2)4COF (438 g, yield 88%), containing ~20% ICF2CF2I. The product was used in the subsequent step without further purification.
19F NMR, δ : 23.6 (t, 6 Hz, 1F, COF), -60.1 (t, 14 Hz, 2F, ICF2), -113.9 (m, 2F, CF2COF), -119.4 (q, 11 Hz, 2F, CF2CF2), -122.9 (m, 2F, CF2CF2).
1-Iodo-5-methoxyperfluoropentane (5)
To a stirred suspension of calcined KF (68 g, 1.17 mol) in anhydrous diglyme (500 mL) at ~15°С, I(CF2)4COF (400 g, 0.86 mol based on the pure product) containing ~20% of ICF2CF2I, was added. The resulting mixture was stirred at room temperature for one hour. Then (MeO)2SO2 (147 g, 1.17 mol) was added at 30 – 35°С and the mixture was stirred at 50 – 60°С for 3 h. The mixture was cooled to room temperature, poured into cold 5% hydrochloric acid (1 L), the bottom layer was washed twice with 500 mL of 5% hydrochloric acid each and distilled to give I(CF2)5OCH3 (280 g, yield 80%), bp 65С (10 Torr).
1H NMR, δ : 3.5 (s, OCH3).
19F NMR, δ : -59.4 (s, 2F, ICF2), -89.5 (s, 2F, CF2OCH3), -114.2 (s, 2F, CF2CF2), -122.4 (s, 2F, CF2CF2), -126.3 (s, 2F, CF2CF2OCH3).
(5-Methoxyperfluoropentyl)trimethyl silane (6)
To a stirred suspension of Zn powder (41.6 g, 0.636 mol) and CuCl (3.2 g, 0.032 mol) in anhydrous DMF (200 mL), ClSiMe3 (3.2 g, 0.03 mol) was added and the resulting mixture was stirred for 15 – 20 min. After that, CH3SO3SiMe3 (90.4 g, 0.54 mol) was added dropwise at 5 – 10°С and then I(CF2)5OCH3 (200 g, 0.49 mol) was added dropwise at 11 – 13°С. The mixture was stirred at room temperature for 30 min, transferred to a separatory funnel, and allowed to separate into layers. The upper layer was separated to give Me3Si(CF2)5OCH3 (156 g, ~80% yield) with an admixture of DMF, Me3SiOSiMe3 and a small amount of H(CF2)5OCH3. The product was used in the subsequent step without further purification.
1H NMR, δ : 3.9 (s, 3H, OCH3), 0.5 (s, 9H, Si(CH3)3).
19F NMR, δ : -89 (s, 2F, CF2OCH3), -119.8 (s, 2F, CF2CF2), -122.7 (s, 2F, CF2CF2), -125.9 (s, 2F, CF2CF2OCH3), -128.7 (t, 17 Hz, 2F, CF2SiMe3).
5-Methoxyperfluoropentanesulfonyl fluoride (7)
To a stirred suspension of calcined KF (27 g, 0.46 mol) in anhydrous DMF (230 mL), a solution of SO2 in DMF (27.8%, 108 g, 0.46 mol of SO2) was added while cooling with cold water. After that, Me3Si(CF2)5OCH3 (150 g, ~0.38 mol), obtained as described above, was added at 15 – 20°С and the mixture was stirred at room temperature for several hours. Next, Cl2 (65 g, 0.9 mol) was bubbled into the reaction mixture with vigorous stirring at 0 – 5°С. The resulting mixture was poured into an equal volume of cold 5% hydrochloric acid; the bottom layer was separated, washed with 5% hydrochloric acid to give ClSO2(CF2)5OCH3 (112 g), which was further mixed with pre-melted sulfolane (200 mL) and then calcined KF (48 g, 0.83 mol) was added to the resulting mixture with stirring. The mixture was stirred at room temperature for 3 h, poured into cold 5% hydrochloric acid (400 mL), the bottom layer was separated, washed with an equal volume of 5% hydrochloric acid each and distilled to give FSO2(CF2)5OCH3 (77 g, yield 55%), bp 143 – 144оС.
1H NMR, δ : 3.9 (s, OCH3).
19F NMR, δ : 44.3 (s, 1F, SO2F), -90 (s, 2F, CF2OCH3), -109.1 (s, 2F, CF2SO2F), -121.4 (s, 2F, CF2CF2), -123.2 (s, 2F, CF2CF2), -126.6 (s, 2F, CF2CF2OCH3).
5-(Fluorosulfonyl)perfluoropentanoyl fluoride (8)
To stirred SbCl5 (6.3 g, 0.021 mol), FSO2(CF2)5OCH3 (77 g, 0.21 mol) was added dropwise at 80°C at such a rate that the reaction mixture slightly boils. The mixture was heated to reflux for one more hour and distilled, collecting a fraction boiling 100°C. Subsequent distillation gave FSO2(CF2)4COF (55 g, yield 80%), bp 88 – 90°С.
19F NMR, δ : 44.9 (m, 1F, SO2F), 22.9 (t, 5 Hz, 1F, COF), -109.3 (t, 11 Hz, 2F, CF2SO2F), -119.4 (s, 2F, CF2COF), -121.1 (s, 2F, CF2CF2), -123.3 (s, 2F, CF2CF2).
5-(Fluorosulfonyl)perfluoropentyl vinyl ether (9)
To a stirred suspension of calcined KF (8.8 g, 0.15 mol) in anhydrous diglyme (300 mL) at 5 – 10°С, FSO2(CF2)4COF (50 g, 0.15 mol) was added. The resulting mixture is stirred at room temperature for an hour. Hexafluoropropene oxide (25 g, 0.15 mol) was then condensed into the stirred reaction mixture at -30°С; the temperature of the resulting mixture was slowly raised to room temperature and the stirring was continued for one hour. After distillation in vacuo (10 Torr), the fraction boiling at 30 – 60°C was collected, which was a mixture of FSO2(CF2)5OCF(CF3)COF and diglyme. This fraction was then added to a stirred suspension of calcined Na2CO3 (15.9 g, 0.15 mol) in anhydrous diglyme (50 mL) at 55 – 75°C at a rate such that the gas evolution was not too intense. At the end of the gas evolution, the temperature of the reaction mixture was raised to 130 – 140°C and the mixture was stirred at this temperature until the gas evolution was complete. Then the reaction mixture was cooled to room temperature, poured into cold 5% hydrochloric acid (500 mL), the bottom layer was separated, washed several times with water and distilled to give FSO2(CF2)5OCF=CF2 (41 g, 65%), bp 135 – 136°С.
Aknowledgements
The study was performed within the framework of the State Assignment of the Ministry of Science and Higher Education of the Russian Federation (Theme No. 075-00277-24-00) using the equipment of the Center for Molecule Composition Studies of INEOS RAS. Financial support of SISTEMA Public Joint Stock Financial Corporation is gratefully acknowledged.
References
- W. Grot, Fluorinated Ionomers, Second Edition, Elsevier Inc, 2011.
- R. Souzy, B. Ameduri, Prog. Polym. Sci., 2005, 30, 644-687.
- M. Odgaard, The Use of Per-Fluorinated Sulfonic Acid (PFSA) Membrane as Electrolyte in Fuel Cells, In: T. Nakajima, H. Groult (eds.), Advanced Fluoride-Based Materials for Energy Conversion. Elsevier Inc, 2015.
- T. Hirai, Y. Morizawa, Fluorinated Ionomers and Ionomer Membranes: Monomer and Polymer Synthesis and Applications, In: B. Ameduri, H. Sawada (eds.) Fluorinated Polymers, Volume 2: Applications, RSC Polymer Chemistry Series, 24, 2017.
- S.J. Hamrock, M.A. Yandrasits, J. Macromol. Sci., Part C, 2006, 46, 219-244.
- C.M. Orsino, Influence of Sidechain Structure and Interactions on the Physical Properties of Perfluorinated Ionomers, Dissertation submitted to the faculty of the Virginia Polytechnic Institute and State University, 2020.
- O.N. Primachenko, E.A. Marinenko, A.S. Odinokov, S.V. Kononova, Y.V. Kulvelis, V.T. Lebedev, Polym. Adv. Technol., 2021, 32, 1386-1408.
- N. Zhao, Z. Shi, F. Girard, Materials, 2022, 15, 78.
- D. Madhav, J. Wang, R. Keloth, J. Mus, F. Buysschaert, V. Vandeginste, Energies, 2024, 17, 998.
- CN107298647B (2019).
- J. Peng, X. Fan, G.A. Goenaga, C.M. Weiss, C.A. Neal, N. Cantillo, T.A. Jr. Zawodzinski, ACS Appl. Polym. Mater., 2023, 5, 9940-9951.
- S.M. Igumnov, A.A. Tyutyunov, RU2475477C1 (2013).
- M. Emery, M. Frey, M. Guerra, G. Haugen, K. Hintzer, K.H. Lochhaas, P. Pham, D. Pierpont, M. Schaberg, A. Thaler, ECS Trans., 2007, 11, 3-14.
- D.F. Mukhametshin, A.A. Deryabin, M.M. Tatarenkova, RU2800857C1 (2023).
- C.G. Krespan, D.V. England, J. Am. Chem. Soc., 1981, 103, 5598-5599.
ARTICLE INFO
Received 07 June 2024
Accepted 26 June 2024
Available online June 2024
Recommended for publication by PhD M.A. Manaenkova
eLIBRARY Document Number (EDN) YWSFGA

Fluorine Notes, 2024, 154, 3-4
